Monitoring of Biological Matrices by GC–MS-MS for Chemical Warfare Nerve Agent Detection
Special Issues
A fluoride-regeneration approach enables biomonitoring of chemical warfare nerve agents.
Traditionally, an indication of exposure to chemical warfare nerve agents (CWNAs) is a depression of cholinesterase activity. However, this method is nonspecific for etiology and source and generally cannot differentiate between past and current exposures. We have evaluated the biomonitoring of CWNAs in various biological matrices from guinea pigs using the technique of fluoride ion regeneration followed by GC–MS-MS analysis.
Although the use of chemical warfare nerve agents (CWNA) such as sarin (O-isopropyl methylphosphonofluoridate, military designation GB), cyclosarin (O-cyclohexyl methylphosphonofluoridate, GF), and VX (O-ethyl-S-diisopropylaminoethyl methylphosphonothioate) (see Figure 1) is forbidden by the Chemical Weapons Convention, documented cases of the use of these nerve agents exist (1–3). The extreme toxicity of these nerve agents makes retrospective detection of potential CWNA exposure an important issue both for the civilian population and military personnel. When exposure is sufficient, clinical symptoms observed include salivation, miosis, tremors, and death (4). These physiological effects result from inhibition of the cholinesterase enzymes distributed throughout the nervous system with a resultant build up of the neurotransmitter acetylcholine. However, when exposure occurs at a very low level, few symptoms, if any, may be present. Therefore, assessing exposure is done typically by means of biomonitoring, a measurement of the internal dose for biological monitoring of exposure.


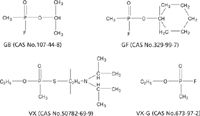
Figure 1: Structures of CWNAs.
Traditionally, an indication of exposure to nerve agents is a depression of cholinesterase activity, specifically erythrocyte acetylcholinesterase (AChE), which has been shown to correlate with the severity of symptoms of CWNA poisoning at high exposure levels. However, this method is nonspecific for etiology and source and generally cannot differentiate between past or current exposures. Furthermore, because of the large inter- and intra-individual variation of erythrocyte AChE activity, a marginal inhibition is impossible to assess unless pre-exposure values have been determined. We have evaluated an alternate method of biomonitoring.
The method reported here employs the fluoride reactivation or regeneration approach first described by Polhuijs and colleagues (1), wherein the corresponding phosphonofluoridate (GB or GF) is regenerated from an inhibited phosphonyl moiety after the addition of fluoride ions. For the VX exposures, biomonitoring employs the fluoride-induced reactivated phosphonyl moiety of VX, O-ethyl methylphosphonofluoridate, or VX-G (5–7), also shown in Figure 1. The VX-G, GB, GF, and deuterated analogs were isolated by means of solid phase extraction followed by analysis using isotope-dilution gas chromatography–tandem mass spectrometry (GC–MS-MS).
Experimental
Animal Exposures: The animal model of choice for this study was the male Duncan-Hartley guinea pig, weighing 250–350 g (Charles River Labs, Kingston, New York). Whole-body inhalation exposures (GB and GF) were conducted in a 1000-L dynamic airflow Rochester-style inhalation chamber (8). Animals were exposed for 60 min to either 1.6 mg/m3 for GB or 1.47 mg/m3 for GF corresponding to 0.4 × LCt50. Before removing the animals, the chamber was purged with air for 10 min after completion of agent exposure. For percutaneous exposures, a 60-μg/kg (0.4 × LD50) 10% VX, 90% isopropanol dosage was applied to the left flank of the animals where their fur had been clipped previously using a 1-μL Hamilton syringe (Hamilton Company, Reno, Nevada). Immediately after agent application, a plastic cap was placed over the exposure site and held in place by double-sided carpet tape. A self-adhesive elastic bandage was wrapped several times around the animal to cover the exposure site, carpet tape, and plastic cap. At 6 h after agent application, the bandage was cut and the carpet tape and plastic cap were removed, exposing the application site. All studies were approved by the U.S. Army Medical Research Institute of Chemical Defense Institutional Animal Care and Use Committee and in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care International. Carotid artery blood draws (0.5–1.0 mL) were taken before the start of the exposure and at periodic intervals throughout into BD Vacutainer tubes (BD, Franklin Lakes, New Jersey) containing K2EDTA and stored at 4 °C until analysis. Tissue samples were stored at –70 °C. During the study, the guinea pigs were observed for clinical manifestations of agent toxicity, but none were observed.
Sample Preparation: For the blood assays, the target analyte was extracted using either Sep-Pak C18 or Oasis HLB solid-phase extraction (SPE) cartridges (both from Waters Corp., Milford, Massachusetts) following a pH adjustment to pH 3.5 with a sodium acetate–acetic acid buffer and the addition of potassium fluoride. This induces the formation of the phosphonofluoridate from either protein bound or free agent. A deuterated internal standard also was added before the solid-phase extraction for quantitation. The analytes and internal standards were eluted with ethyl acetate. Tissue sample preparation also employed freeze-fracture pulverization under cryogenic temperatures using a Tissue Cryoprep system and an S-series focused acoustic energy system (Covaris, Woburn, Massachusetts).


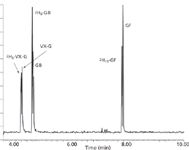
Figure 2: Total ion chromatogram of 200-pg standard.
Instrumentation: Analyses were performed using an Agilent Technologies (Santa Clara, California) model 6890 gas chromatography (GC) system interfaced to a Waters Micromass Quattro micro GC tandem quadrupole mass spectrometer. GC separations were achieved using a 30 m × 0.25 mm, 0.25-μm df Restek Rtx-5MS column (Bellefonte, Pennsylvania). The carrier gas was helium with a flow rate of 1 mL/min. Injections of 1.0 μL were made by autoinjector into a splitless injector port at a temperature of 250 °C. The initial oven temperature of 35 °C was held for 1 min, then ramped at 15 °C/min to 100 °C, and ramped again at 35 °C/min to 280 °C and held for an additional 1 min. Typical elution times are shown in Figure 2.


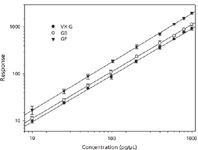
Figure 3: Calibration curves for CWNAs.
Samples were ionized by positive-ion chemical ionization (CI) with ammonia reagent gas. CI source conditions were optimized using Fluoroether E3 tuning compound with methane reagent gas. Mass spectra were obtained at a dwell time of 0.05 s for each transition in the multiple reaction monitoring (MRM) mode. Argon was used as the collision gas at a pressure of ~2 mTorr, with a collision-induced dissociation (CID) energy of 10 eV. The following transitions were monitored for the respective analytes: m/z 144 > 99 for VX-G, m/z 149 > 100 for 2H5-VX-G, m/z 158 > 99 for GB, m/z 164 > 100 for 2H6-GB, m/z 198 > 99 for GF, and m/z 209 > 100 for 2H11-GF.


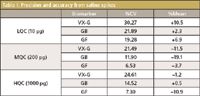
Table I: Precision and accuracy from saline spikes
Results
Method Validation: Figure 3 shows the response of the chemical warfare nerve agents to the internal standards as a function of picograms agent injected onto the column (for a 1-μL injection) for a series of calibration standards prepared in ethyl acetate. The data are an average taken over several weeks and show good correlations over two orders of magnitude (VX-G, r2 = 0.9995, GB, r2 = 0.9997, GF, r2 = 0.9987). The precision (presented as the coefficient of variation or CV) and accuracy (or mean) of the assays were determined by replicate analyses (n = 5) of multiple aliquots of saline spiked at three different concentrations. These concentrations represented a low quality control (LQC), a medium quality control (MQC), and a high quality control (HQC) equivalent to the concentrations of biomarkers on column. The results are summarized in Table I. Recoveries of the biomarkers were measured by spiking blood with either VX, GB, or GF at the equivalent of 0.1 nmol of agent per gram of blood and assaying for the respective analyte. Results varied from 60% to 80%.


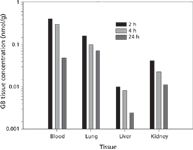
Figure 4: Concentration of GB in various tissues as a function of time post-exposure.
Sample Analyses: Figure 4 shows the concentrations of regenerated sarin (GB) determined in various tissues as a function of time following the 60-min inhalation exposure. As expected, once the nerve agent enters the systemic circulation, it is distributed throughout the tissues of the body. Each value represents the average of six or seven individual determinations. As the data indicate, there is a definite decrease in the concentration of GB with time. Based on additional concentration/time measurements (not shown), all four biological matrices exhibit typical first-order elimination kinetics of the form c(t) = ae–kt, where half-lives for regenerated sarin were estimated as blood, 3.4 h; lung, 22 h; liver, 11 h; and kidney, 14 h. Figure 5 shows the same plot for the concentrations of regenerated cyclosarin (GF) as a function of time following a similar 60-min inhalation exposure. In general, the data are similar to the GB exposure with comparable half-lives for regenerated GF.


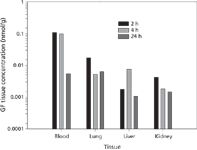
Figure 5: Concentration of GF in various tissues as a function of time post-exposure.
Figure 6 shows the concentrations of VX-G analog in various tissues as a function of time following a percutaneous exposure to VX. These data clearly show the effect of the dermal exposure route. Although blood levels remain essentially unchanged, VX-G concentrations in the three target organs actually increase with time. As the data indicate, there is a significant amount of VX-G recovered in the skin at the site of exposure even 24 h later. A spontaneous release of this agent from bound sites in the skin with a systemic circulatory uptake might account for the observed target organ increases. This mode of action could impact postexposure decontamination and treatment efforts.


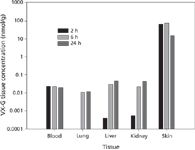
Figure 6: Concentration of VX-G in various tissues as a function of time post-exposure.
Acknowledgments
The authors wish to acknowledge Drs. Christopher Whalley and Lucille Lumley for their technical assistance as Principal Investigators, and all the members of the Analytical Toxicology Branch for their help with the regeneration assays. This research was supported by the Defense Threat Reduction Agency. DTRA neither endorses nor recommends the use of any products, techniques, or materials mentioned or pictured here.
References
(1) M. Polhuijs, J.P. Langenberg, and H.P. Benschop, Toxicol. Appl. Pharmacol. 146, 156–161 (1997).
(2) M. Minami, D.M. Hui, M. Katsumata, H. Inagaki, and C.A. Boulet, J. Chromatogr. B 695, 237–244 (1997).
(3) H. Tsuchihashi, M. Katagi, M. Nishikawa, and M. Tatsuno, J. Anal. Toxicol. 22, 383–388 (1998).
(4) F.R. Sidell, J. Appl. Toxicol. 14, 111–113 (1994).
(5) E.M. Jakubowski, L.S. Heykamp, H.D. Durst, and S.A. Thomson, Anal. Lett. 34, 727–737 (2001).
(6) C.E.A.M. Degenhardt, K. Pleijsier, M.J. van der Schans, J.P. Langenberg, K.E. Preston, M.I. Solano, V.L. Maggio, and J.R. Barr, J. Anal. Toxicol. 28, 364–371 (2004).
(7) J.M. McGuire, J.T. Taylor, C.E. Byers, E.M. Jakubowski, and S.A. Thomson, J. Anal. Toxicol. 32, 73–77 (2008).
(8) C.E. Whalley, J.M. McGuire, D.B. Miller, E.M. Jakubowski, R.J. Mioduszewski, S.A. Thomson, L.A. Lumley, J.H. McDonough, and T.-M.A. Shih, Inhalation Toxicol. 19, 667–681 (2007).
Jeffrey M. McGuire, Edward M. Jakubowski, Jr., and Sandra A. Thomson are with the US Army Edgewood Chemical Biological Center, Aberdeen Proving Ground, Maryland.















Mass Spectrometry for Forensic Analysis: An Interview with Glen Jackson
November 27th 2024As part of “The Future of Forensic Analysis” content series, Spectroscopy sat down with Glen P. Jackson of West Virginia University to talk about the historical development of mass spectrometry in forensic analysis.

Detecting Cancer Biomarkers in Canines: An Interview with Landulfo Silveira Jr.
November 5th 2024Spectroscopy sat down with Landulfo Silveira Jr. of Universidade Anhembi Morumbi-UAM and Center for Innovation, Technology and Education-CITÉ (São Paulo, Brazil) to talk about his team’s latest research using Raman spectroscopy to detect biomarkers of cancer in canine sera.



