Online Automated SPE–LC–MS-MS Quantitation of Sulfonamide Antibiotics and Pesticides in Surface Waters Using a Triple-Quadrupole Mass Spectrometer and Electrospray Ionization Probe
Special Issues
This article describes a fully automated online solid-phase extraction–liquid chromatography–tandem mass spectrometry (SPE–LC–MS-MS) setup using a mass spectrometer and an electrospray ionization probe for analyzing different groups of polar contaminants in natural waters. The goal was to develop an online SPE method for the quantification of sulfonamide antibiotics, including their acetyl metabolites, as well as for frequently used pesticides (triketones, phenylureas, chloracetanilides, phenoxyacetic acids, amides, and triazines) in ambient waters. The analytical methods were applied successfully for a field study in an agricultural region within the catchment area of Lake Greifensee near Zurich, Switzerland.
As an aquatic research institute committed to the ecological, economical, and socially responsible management of water, the work of EAWAG — Swiss Federal Institute of Aquatic Science and Technology (Dübendorf, Switzerland) — focuses on creating a link between science and practical application. The organization works on the philosophy that clean waters are not self-evident. EAWAG's task as the Swiss national research center for water pollution control is to ensure that concepts and technologies pertaining to the use of natural waters are continuously improved.
While traditional sample preparation for organic compounds using offline solid-phase extraction (SPE) has many advantages, the goal was to develop a setup for online SPE coupled with liquid chromatography (LC) and tandem mass spectrometry (MS-MS) — SPE–LC–MS-MS — to combat some of the disadvantages. The most urgent of the disadvantages with offline SPE was the length of time needed to work up the large number of samples typically analyzed in EAWAG's studies. Further advantages expected with an online SPE method included the direct coupling of the extraction to the mass spectrometer to allow unattended 24/7 operation, and the storage of the extraction method within the raw data files, thereby cutting down the laboratory's administrative overhead.
In keeping with EAWAG's objectives regarding natural waters, bioactive compounds common to agriculture and private households were water contaminants of particular interest because of their potentially unwanted side effects to humans and aquatic organisms. Antibiotics enter the environment as a result of the practice of spreading antibiotic-containing manure in agriculture (1) or by input from wastewater treatment plants after excretion by humans (2). Pesticides are introduced into the environment intentionally for crop protection in agricultural areas or for nonagricultural use in urban areas. Even though modern pesticides are fairly degradable, surface waters can contain high concentrations due to losses from agricultural land or direct input from point sources.
These compounds have low octanol–water partitioning coefficients (logKow < 3) and high water solubilities (milligrams per liter to grams per liter) because of their functional groups with H-donor–acceptor properties. Additionally, most of them show pKa values in the environmentally relevant range and are typically anionic in natural waters. The input of these substances from diffuse and point sources to surface water is highly dynamic (3). Therefore, the requirements for this study include high sample throughput and a dynamic measuring range over several orders of magnitude for the reliable quantification of the load or the concentration dynamic of these substances in catchment studies for mass balance or risk assessment purposes.
To get accurate mass balances also during low flow situations in rivers and streams, analytical methods must exhibit sensitivity in the low nanograms-per-liter range. In addition, high selectivity is required to avoid interference by matrix constituents. Presently, LC–MS-MS is the method of choice for this type of analysis. A major advantage of using LC instead of gas chromatography (GC) is that derivatization of polar analytes is not required (4). However, water samples usually must be preconcentrated before analysis, which is typically done by time-consuming and costly offline SPE.
The simplest approach to automation generally is a single cartridge approach (5). The direct coupling of SPE to LC eliminates several working steps, such as evaporation, reconstitution, and injection. This results in a faster and more precise procedure since the total enriched amount of substance is eluted directly to the liquid chromatograph (6). In addition, automated online SPE has the potential to reduce procedural errors. In contrast to pharmacological studies, where cleanup is often the main issue, achieving quantifiable analyte amount is usually the major challenge in environmental analysis. Sample volumes of a few tens of milliliters often have to be enriched to quantify analytes in the low nanogram-per-liter range with conventional LC–MS-MS systems, which typically have absolute sensitivities of approximately 0.5–10 pg.
Online SPE methods that use manual loop injections (7) or an LC pump (8) for sample delivery are not compatible for routine analysis. Applications designed for multiple sample handling without an autosampler either use a multiport valve (9) or a solvent delivery system (10–12). The main drawback of these systems for routine analysis is the limited number of individual samples that can be processed. Generally, a maximum of 22 samples can be analyzed without manual interaction by combining several valves (13). This limitation would prevent unattended analysis of large sample sets over several days (such as during weekends). Therefore, this study presumes that an autosampler, which can handle sample volumes up to 10 mL and more than 50 samples per sequence, is a prerequisite for high sample throughput in routine analysis.
Experimental
Instrumentation
The study used a tridirectional autosampler with an 80-μL side-port syringe (80-mm needle) combined with a large volume dispenser module (10-mL dispenser syringe with a 10-mL loop) and two sample trays with 64 positions for 20-mL vials for sample injection and buffer addition. Sample enrichment was achieved with an 18-mL sample loop on a 20 mm × 2.1 mm hydrophilic–lipophilic balanced (HLB) extraction cartridge (25 μm particle size) using two six-port valves. HPLC-grade eluents were used.
The LC pump system consisted of a binary Thermo Scientific Surveyor LC pump (load pump), a quaternary LC pump (elution pump), an isocratic LC pump (precolumn addition pump), and a column oven. A 125 mm × 2 mm, 5-μm particle size column was used with a guard column for the chromatographic separation.
The liquid chromatograph was coupled with an Ion Max compatible heated electrospray ionization (H-ESI) probe to a TSQ Quantum Ultra triple-quadrupole mass spectrometer (Thermo Fisher Scientific), operated under unit resolution in the selected reaction monitoring (SRM) mode. Details of the substance-specific parameters for the ionization and detection of the pesticides are available in reference 14, and the sulfonamides are published in reference 15, along with detailed methodology.
Setup
The method setup used an online SPE–LC coupling with two switching valves. The dispenser system consisted of a large-volume dispenser syringe connected to the autosampler syringe and to a wash solution via dispenser valve. An additional loop was inserted between dispenser valve and autosampler syringe to avoid contamination of the dispenser syringe. Sample enrichment was performed by the load pump, which was also used for washing and conditioning the extraction cartridge. The load pump was connected to the 18-mL loop via valve 1. Valve 1 was linked with valve 2, where the elution pump and the extraction cartridge were attached. The precolumn addition pump was placed between valve 2 and the analytical column using a mixing tee. The whole procedure was controlled through Xcalibur software version 1.4 (Thermo Fisher Scientific).
The online SPE procedure consists of three main steps: loading, enrichment, and elution. The 18-mL sample loop was loaded with two times 9.5 mL of sample. The sample was enriched with a flow rate of 2 mL/min. Elution was done in the back-flush mode. The SPE eluate was mixed with buffered water from the precolumn addition pump before the analytical column. The high pressure gradient for the analytical separation was achieved by changing the ratio of the elution pump (eluents A and B) and the precolumn addition pump (eluent C). Different solvents were used for the eluents A, B, and C in the three different analytical methods. The composition of the eluents and the gradients for the sulfonamid and the neutral pesticides are shown in Figure 1. By increasing the flow of the precolumn addition pump during the first 4 min (Figure 1a, solvent C), a sufficient trapping of the very polar sulfonamides on the analytical column could be realized. The LC program for the acidic pesticides is only slightly different from that of the neutral pesticides (15). The cleaning of the sample loop as well as the cleaning and conditioning of the SPE cartridge was done by subsequent rinsing steps of water and acetonitrile with the loading pump.


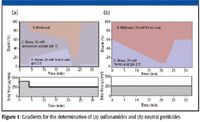
Figure 1
Sample Preparation
The study involved mass flux studies of veterinary sulfonamide antibiotics on grasslands and pesticides in crop protection during the spring and summer of 2003 in the basin of Lake Greifensee, near Zurich, Switzerland. An automatic water sampling station was installed in the creek at the outlet of a subcatchment of 0.7 km2 — an area of intensive agricultural production, mainly grasslands and crop production. Catchment discharge volumes were measured and flow-proportional water samples were taken at very high frequency during the whole investigation period. The surface water samples for extraction recovery determinations were collected from the outflow of Lake Greifensee on 22 January 2003 and from the creek at the outlet of the investigated subcatchment on 26 February 2003. All samples were transferred to 1 L glass bottles and stored in the dark at 4 °C for a maximum of 6 months until analysis; storage stability was proven by repetitive analysis of one fortified sample.
Samples were filtered at room temperature in the laboratory with a 250-mL bottle-top filtration unit, using 50-mm diameter, 0.45-μm pore size cellulose nitrate membrane filters. Isotope-labeled internal standards for most of the analytes were spiked to the samples via the autosampler, to a concentration level of 50 ng/L. Filtration recoveries — validated using fortified lake and creek water samples — were higher than 95% for all substances. For reproducible trapping on the extraction cartridge, the sample pH (ranging from 6.5 to 8.5) was adjusted to 4 by adding 80 L of 5M acetate buffer (composition: 4:1 [v/v] 5 M acetic acid–5 M sodium acetate) via the autosampler. This yielded a concentration of 20 mM acetate in the sample (nanopure, creek lake, and groundwater), which was sufficient for adequate buffering of the different environmental water samples.
Validation
The following parameters were determined during validation of the three analytical methods: absolute extraction recovery, matrix effects, limits of quantification (LOQ) and limits of detection (LOD), linearity, precision, and accuracy. Absolute extraction recovery was determined in nanopure water and natural surface water at six concentration levels (100, 250, 500, 1000, 2500, and 5000 ng/L). Therefore, the SPE elution step (10 min) was collected, spiked with internal standard solution, and measured by 20-μL loop injection without further pre-concentration to avoid another pathway of potential losses and quantified with standards in nanopure water (external calibration, levels 2, 5, 10, 20, 50, and 100 ng/mL). The absolute extraction recovery of each analyte resulted from calculating the ratio between the slope of the extracted calibration curve (nanopure or matrix) and the slope of the external calibration. Additionally, breakthrough samples were collected by sampling the waste line of valve 2 during enrichment.
Results and Discussion
The efficiency and applicability of the developed setup was demonstrated with three analytical methods for the sulfonamides sulfadiazine, sulfadimethoxine, sulfamethazine, sulfamethoxazole, and sulfathiazole including their acetyl metabolites; the neutral pesticides atrazine and its desethyl metabolite, dimethenamide, diuron, isoproturon, metolachlor, simazine, tebutam, and terbuthylazine; and the acidic pesticides 2,4-D, dimethenamide-ethanesulfonic acid (ESA) and oxanilic acid (OXA), MCPA, mecoprop, mesotrione, metolachlor-ESA, and OXA and sulcotrione. The three analytical methods were applied successfully for a field study of sulfonamide antibiotics, neutral, and acidic pesticides in an agricultural region within the catchment area of Lake Greifensee near Zurich, Switzerland.
The instrumental setup for the online SPE–LC coupling was accomplished by several upgrades to a conventional LC–MS-MS system: a dispenser syringe, two loops, two LC pumps, two six-port valves, and an online extraction cartridge. The required sensitivity of a few nanograms per liter was achieved with enrichment of 18 mL realized by dual injection with the dispenser syringe. The excellent sensitivity of the method is illustrated in Figure 2, which shows the peaks of selected pesticides for a 1-ng/L standard. The combination with the freely programmable autosampler allowed automatic sample buffering and could also be used for reagent addition in future applications. The addition of two large capacity sample trays for 20-mL vials enabled sequences to be executed with large numbers of samples for several days, including overnight without surveillance on-site. For full details on the instrumentation setup, see reference 15.



Figure 2
High extraction recoveries were achieved for all of the compounds: sulfonamides 85–104% (average: 91±5)%, neutral pesticides 95–111% (102±4)%, and acidic pesticides 99–112% (105±3)% (for analyte-specific details, see reference 15). There were no significant differences in extraction recoveries between nanopure and surface water for any of the three analyte groups. The recoveries for the substances without corresponding internal standard were not significantly different (two-sided, heteroscedastic t-test, p > 5%) from the ones with isotope-labeled internal standard within the substance group. No breakthrough was observed for most of the substances, except for the sulfonamides and dimethenamide OXA, where breakthrough <5% of the enriched amount was detected. This is much lower than with the published offline procedure (14). This improvement was ascribed to the 15 times higher sorbent-to-sample volume ratio of the online method compared to offline SPE.
Matrix effects were evaluated during analysis of several hundred environmental samples from a broad range of different matrices (such as creek water from an agricultural area, lake water, and groundwater) by monitoring the change of the area of the isotope-labeled standard, which was spiked at the same concentration in every sample. Full details on matrix effects have been published previously (15).
Sulfamethazine and sulfamethoxazole containing manure as well as pesticides were each applied on several fields with areas of 0.5–1.5 ha in the spring of 2003. An overview of the temporal variation of analyte concentrations measured in the creek is given in Figure 3. In general, the concentrations for each analyte follow discharge dynamics of the creek, which were already shown for neutral pesticides in an earlier study in the same catchment (3). Measured concentrations were highly variable, indicating the need for frequent sampling and sample analysis. Concentration ranged from a few tens of nanograms per liter up to several thousand nanograms per liter after the respective applications. This demonstrates the requirement for a large dynamic measuring range of the analytical method as well as high sensitivity to quantify "base flow concentrations" and "preapplication samples." Furthermore, prevention of cross-contamination is of particular importance in measuring samples with differing concentration by orders of magnitude.



Figure 3
The interday precision of the developed analytical methods was proven by alternating measurement of the samples in a period of more than two months, including different extraction cartridges and different calibration standards. The perfect congruency of the time course of the measured concentrations illustrates the reliability of the employed analytical procedure. In total, 600 surface water samples, 400 standards, and 200 quality control samples were measured during the three-month field study. The high sample throughput of the developed instrumental setup enabled the fast and precise quantification of sulfonamides and pesticides in the highly dynamic creek water system — an absolute must in mass balance studies.
Conclusions
The required high sensitivity and high sample throughput in the low nanogram-per-liter range were achieved using the automated online SPE–LC–MS-MS method with the combination of commercially available standard components. Also, the method reduced manual sample preparation to sample filtration and spiking of the internal standard solution, thereby decreasing the laboratory time by more than a factor of five.
The flexibility of the instrumental setup could support the tailor-made optimization of the substance-specific properties at every step of the procedure: pH and solvent composition can be adjusted for enrichment, elution, separation, and ionization. As a result, the instrumental setup could apply to many other polar substances within a range of physicochemical properties, such as other antibiotic classes, pesticides, or biocides.
Acknowledgments
This work was partly funded by the Swiss National Research Program NRP49 "Antibiotic resistance" (4049-63282) and by the Swiss Agency for the Environment, Forests, and Landscape (SAEFL). The authors are very grateful to Ch. Goetz, M. Ruff, and St. Mueller for their collaboration during the method development.
References
(1) M.Y. Haller, S.R. Muller, C.S. McArdell, A.C. Alder, and M.J.-F. Suter, J. Chromatogr. A 952, 111 (2002).
(2) R. Hirsch, T. Ternes, K. Haberer, and K.L. Kratz, Sci. Total Environ. 225, 109 (1999).
(3) C. Leu, H. Singer, C. Stamm, S.R. Muller, and R.P. Schwarzenbach, Environ. Sci. Technol. 38, 3827 (2004).
(4) M. Petrovic, M.D. Hernando, M.S. Diaz-Cruz, and D. Barcelo, J. Chromatogr. A 1067, 1 (2005).
(5) C.R. Mallet, Z.L. Lu, J. Mazzeo, and U. Neue, Rapid Commun. Mass Spectrom. 16, 805 (2002).
(6) H. Obrist, Chimia 55, 46 (2001).
(7) E. van der Heeft, E. Dijkman, R.A. Baumann, and E.A. Hogendoorn, J. Chromatogr. A 879, 39 (2000).
(8) A. Asperger, J. Efer, T. Koal, and W. Engewald, J. Chromatogr. A 960, 109 (2002).
(9) T.D. Bucheli, S.R. Muller, P. Reichmuth, S.B. Haderlein, and R.P. Schwarzenbach, Anal. Chem. 71, 2171 (1999).
(10) J. Slobodnik, A.C. Hogenboom, J.J. Vreuls, J.A. Rontree, B.L.M. van Baar, W.M.A. Niessen, and U.A.Th. Brinkman, J. Chromatogr. A 741, 59 (1996).
(11) A.C. Hogenboom, P. Speksnijder, R.J. Vreeken, W.M. Niessen, and U.A.Th. Brinkman, J. Chromatogr. A777, 81 (1997).
(12) R.B. Geerdink, A. Kooistra-Sijpersma, J. Tiesnitsch, P.G.M. Kienhuis, and U.A.Th. Brinkman, J. Chromatogr. A 863, 147 (1999).
(13) S. Rodriguez-Mozaz, M.J.L. de Alda, and D. Barcelo, Anal. Chem.76, 6998 (2004).
(14) L.G. Freitas, C.W. Gotz, M. Ruff, H.P. Singer, and S.R. Muller, J. Chromatogr. A 1028, 277 (2004).
(15) K. Stoob, H.P. Singer, C.W. Goetz, M. Ruff, and S.R. Mueller, J. Chromatogr. A 1097, 138–147 (2005).
Heinz P. Singer and Krispin Stoob are with the Swiss Federal Institute of Aquatic Science and Technology, EAWAG Environmental Chemistry Department, and Swiss Federal Institute of Technology, ETH, Zurich, Switzerland. E-mail: heinz.singer@EAWAG.ch (H.P. Singer).














High-Speed Laser MS for Precise, Prep-Free Environmental Particle Tracking
April 21st 2025Scientists at Oak Ridge National Laboratory have demonstrated that a fast, laser-based mass spectrometry method—LA-ICP-TOF-MS—can accurately detect and identify airborne environmental particles, including toxic metal particles like ruthenium, without the need for complex sample preparation. The work offers a breakthrough in rapid, high-resolution analysis of environmental pollutants.




Trending on Spectroscopy: The Top Content of 2024
December 30th 2024In 2024, we launched multiple content series, covered major conferences, presented two awards, and continued our monthly Analytically Speaking episodes. Below, you'll find a selection of the most popular content from Spectroscopy over the past year.


