Speciation of Mercury in Crude Oil Using Speciated Isotope Dilution Mass Spectrometry
ICP-MS and IDMS for crude oil analysis are explored.
Mercury, in a variety of chemical and physical forms, is contributed to the environment from many sources, both natural and anthropogenic. It exists naturally in mineral form, usually associated with ores and other geological materials. Mercury enters and is recycled in the environment from a variety of sources, including both natural and anthropogenic (1). The anthropogenic activities are of the greatest importance in the mobilization of this metal and its compounds. Among the contributors of mercury to the environment, the burning of fossil fuel is a significant concern. While many scientific studies have focused upon coal and coal-fired power plants as industrial sources of mercury in the environment, other sources containing different species and concentrations also have generated interest. A deeper understanding of the origins of these species can be obtained by measuring speciated components of this reactive heavy metal. Other contributing sources include natural gas, gas condensate, crude oil, and petroleum, which also can contain significant amounts of mercury (2). Depending upon the region, crude oil produced around the world can contain varying levels of mercury and can range from 0.1 to 20,000 μg/kg (3).
The nature of mercury compounds in crude oil is not well-specified and quantified. Based upon previous studies on its chemical reactivity, the mercury in crude oil has been identified mostly as metallic with trace amounts of inorganic content (for example, mercuric chloride) and organometallics, including, monomethylmercury, monoethylmercury, dimethylmercury and diethylmercury, as well as nonsoluble mercury, such as mercury sulfide and mercury selenide (4). The content and concentration of different species of mercury in crude oil cause processing and environmental concerns. Accurate measurement of major species of mercury is therefore highly desirable for all stakeholders in the fossil fuel continuum.
Determination of mercury species in crude oil is difficult due to the volatile nature of many mercury species and complexity of the matrix. The traditional hot acid digestion procedures could cause lower recovery or transform mercury species during sample preparation. Therefore, an improved sample preparation protocol necessitates different strategies, such as sample preparation in a closed vessel microwave system followed by element-selective determination in the resulting aqueous medium (2).


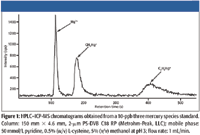
Figure 1
From the literature, it was found that popular techniques for the determination of total mercury use cold vapor atomic absorption spectrometry (AAS) (2) or atomic fluorescence spectroscopy (AFS) (5) and isotope dilution cold vapor inductively coupled plasma–mass spectrometry (ID-CV-ICP-MS) (6). Mercury speciation analysis in crude oil matrices mainly consists of combinations of hyphenated chromatography for separation such as gas chromatography (GC) (7,8) and high performance liquid chromatography (HPLC) (9) with different detectors and detection methods. The detection methods used in GC for the determination of mercury species are ICP-MS (7), AFS (5), atomic absorption spectrometry (AAS) (9), electron-capture detection (ECD) (10), atomic emission spectrometry (AES) (11). The main problem of using GC-based separation in conventional ways is that the analyte must be volatile and thermally stable. To produce a volatile and stable mercury compound, the analyte must be treated with a suitable derivatizing reagent to make sure all the mercury compounds present in the extracted solvent are derivatized completely and back-extracted into the organic solvent, which are then injected into the GC system or extracted on a solid-phase microextraction sorbent in the headspace. Although HPLC separation can be used without derivatization, this method suffers from high detection limits. Many crude oil samples contain low concentrations of mercury species of interest.
The method reported in this article is an improvement of the sample preparation and detection inadequacies and represents a significant progress in the determination of mercury species in crude oil: the closed vessel microwave digestion system completely dissolves the difficult matrix without loss of analyte, and traditional corrections for analyte recovery are not necessary because the isotopically enriched analogues of the species are spiked before extraction–digestion and are equilibrated with the corresponding mercury species present in the sample during extraction to yield a fixed isotope ratio. This ratio is unaffected by any subsequent postequilibration loss of mercury species. Speciated isotope dilution mass spectrometry (SIDMS), the underlying metrology in EPA Method 6800, was used as a diagnostic tool and analytical determinative technique in this study. The unique SIDMS capability performed a dual role of quantifying and correcting for species transformation of up to three or four species simultaneously. In this case, SIDMS was used as a protocol step-evaluation tool to trap and evaluate errors from specific procedural steps. Thermally or chemically caused species interconversions occurring postspiking are traceable using the isotope enrichments in the species and can be corrected quantitatively by monitoring the values of the enriched species-isotopes equilibrated with each sample species. The SIDMS protocol enabled accurate correction for species transformations and determination of species concentrations at the point of spiking. To optimize the protocol, the sample was spiked both before and after its extraction to assess transformation during critical procedural steps. SIDMS was used to identify procedure-altered species distributions in a multistep protocol, and as importantly, to minimize and correct for these species introversions that would have otherwise invalidated our results.
Experimental
Reagents and Chemicals
High-purity double deionized (DDI) water (18 MΩ/cm) was prepared from a Barnstead NANOpure Ultrapure water system (Dubuque, Iowa) and used in the preparation of all solutions throughout this study. Reagent-grade nitric acid, hydrogen peroxide, L-cysteine, pyridine, and methanol were obtained from Fisher Scientific (Pittsburgh, Pennsylvania). Nitric acid was purified in-house prior to use by sub-boiling distillation of the reagent-grade acids in a quartz still (Milestone, Monroe, Connecticut).
Sample and Standards
The crude oil samples used during this study were obtained from and were speciated in collaboration with a major international oil company. The Method 6800-compliant, 3Hg-SPC mercury speciation analysis kit (Applied Isotope Technologies, Sunnyvale, California) containing both natural (isotopic) abundant and isotopic enriched inorganic mercury (199 HgCl2) (91.95%), both naturally abundant and isotopic enriched monomethyl mercury chloride (CH3200 HgCl) (96.41%), and both naturally abundant and isotopic enriched monoethyl mercury chloride (C2H5201 HgCl) (98.11%), and SIDMS-deconvolution software keyed to these isotopic abundances were obtained from Applied Isotope Technologies, Inc. (AIT, Sunnyvale, Califonia). All stock solutions were stored in amber glass bottles in a cold room at 4 °C. Working standards were made daily by dilution with DDI water.
Caution: Mercury compounds, especially monomethyl mercury, are highly toxic materials. Proper knowledge and safety guidelines regarding working with mercury compounds are required to handle these compounds.
Instrumentation
A laboratory microwave system (Ethos 1: Advanced microwave digestion system, Milestone), equipped with temperature and pressure feedback control and magnetic stirring capability was used in this study. This device is accurate in temperature sensing and control to within ± 2.0 °C of set temperatures and automatically adjusts the microwave field output power to achieve preset temperatures. The high-pressure rotor was used with 10 simultaneous extraction vessels per batch. The high-pressure (maximum operating pressure and temperature: 100 bar and 300 °C) closed digestion vessels (model SK10, Milestone) used for both digestions and extractions are made of high purity TFM (a thermally resistant fluoropolymer) and have a capacity of 100 mL each.
Caution: Safety guidelines regarding work with microwave fields in the laboratory must be observed.
The isocratic liquid chromatography system, consisting of a model 818 IC polymeric inert pump, a model 762 software interface (Metrohm Peak, Houston, Texas), and a model 838-advanced sample processor with a 100-μL sample loop was used to automate the sample delivery and sample handling using inert chemical components througout the automation system (Metrohm Peak). A PS-DVB C18 reversed-phase analytical column (150 mm × 4.6 mm, 2 μm) supplied by Metrohm Peak was used in this study to separate inorganic mercury, monomethylmercury and monoethylmercury before ICP-MS analysis. Because no special interface was required between the C18 column and the ICP-MS system, one outlet of the column was directly interfaced to the nebulizer of the ICP-MS system with a piece of PEEK tubing. The other end was connected to a 100-μL sample loop on the sample processor. The chromatography and automation equipment was housed in a high-efficiency particulate air (HEPA) filter class 100 clean hood chamber constructed at Duquesne University (Pittsburgh, Pennsylvania), located adjacent to the ICP-MS system, both located in a class 1000 instrument clean room laboratory. The chromatographic separation was performed as previously described (12) by using isocratic elution with 50 mmol/L pyridine, 0.5% (w/v) L-cysteine, 5% (v/v) methanol at pH 3 at room temperature with a flow rate of 1 mL/min. Before the analysis, the polymeric-based C18 column was preequilibrated with the mobile phase for 30 min.
An HP 4500 ICP-MS system (Agilent Technologies, Palo Alto, California and Yokogawa Analytical System Inc., Tokyo, Japan) was used as isotopic detector for the HPLC system. The sample delivery system consisted of a peristaltic pump and a Scott-type double-pass quartz spray chamber with a concentric nebulizer and a quartz torch. The instrument was fitted with platinum sampler and skimmer cones and was optimized daily using 10 ng/mL tuning solution (Agilent Technologies) containing Li, Y, Ce, and Tl in 0.4% (w/v) L-cysteine (for speciation analysis) and in 1% (v/v) HNO3 (for total mercury analysis). The spectrum mode (direct analysis) was engaged for total mercury analysis: the time resolved analysis (TRA) mode (chromatographic analysis) was engaged for speciation analysis. The ICP-MS operating conditions for total mercury and speciation analyses are provided in Table I.


Table I: Operating conditions for ICP-MS and HPLC
Sample Preparation Procedure
Total Mercury digestion procedure
For total mercury, crude oil samples were digested using EPA Method 3052 (13). To perform the isotope dilution mass spectrometry (IDMS) analysis for the determination of total mercury content in the crude oil samples, approximately 0.5 g of representative aliquots of crude oil samples were weighed into four microwave vessels. Then 0.5 g of 10.0 μg/g 199 Hg2+ was added to each vessel followed by the addition of 9 mL of concentrated HNO3, 2 mL of 30% H2O2, and a magnetic stir bar. The vessels were sealed and digested according to EPA Method 3052. After digestion, the sample aliquots were filtered using a 0.22-mm Millipore glass fiber filter (Fisher Scientific, Pittsburgh, Pennsylvania) and stored at 4 °C until analysis (usually within 48 h).
Mercury species extraction procedure
Extractable Mercury Species: For extractable mercury speciation analysis, the crude oil samples were extracted according to EPA Method 3200 after isotopic labeling (see EPA Method 6800 for SIDMS analysis) (14). Four 500-mg subsamples of the crude oil were weighed into individual precleaned fluoropolymer digestion vessels. A 10-mL volume of 4 M HNO3was placed in each of the microwave extraction vessels. A magnetic stir bar was added to each vessel for thorough mixing of solvent with the sample. The microwave vessels were sealed and irradiated at 100 °C for 10 min with magnetic stirring on. A ramping time of 2 min was used to reach the desired temperature of 100 °C. After microwave irradiation, the vessels were cooled to room temperature and the samples were filtered through a 0.22-mm glass fiber filter (Fisher Scientific) into a 50-mL polypropylene graduated centrifuge tube, diluted with DDI water to 20 mL, and stored in a cold room at 4 °C for less than two days before analysis. Four subsamples of crude oils were prepared in each case. A procedural blank was prepared along with the samples for quality assurance purposes.
Nonextractable Mercury Species
To determine the nonextractable fraction of mercury, the residues left from the "extractable mercury species step" were spiked with isotopically labeled inorganic mercury (199 Hg2+ ) and digested using EPA Method 3052. A volume of 9 mL concentrated HNO3 was placed in each of the microwave extraction vessels. A magnetic stir bar was added to each vessel for thorough mixing of solvent with the sample. The microwave vessels were sealed and irradiated at 180 °C for 10 min with magnetic stirring. A ramping time of 10 min was used to reach the desired temperature of 180 °C. After microwave irradiation, the vessels were cooled to room temperature, filtered through a 0.22-mm glass fiber filter (Fisher Scientific) into a 50-mL polypropylene graduated centrifuge tube, diluted with DDI water to 20 mL, and stored in a cold room at 4 °C for less than two days before analysis. A procedural blank was prepared along with the samples for quality assurance purposes.
Analysis of the extracts by ICP-MS and HPLC-ICP-MS
The prepared solutions were further diluted and analyzed by ICP-MS and HPLC-ICP-MS.
Total mercury determination by ICP-MS
To compare the certified total mercury value with those determined during this study, the digested and extracted mercury solutions were analyzed using ICP-MS in spectrum mode. The plasma parameters used for the analysis are summarized in Table I. Total mercury concentrations were determined by IDMS.
Mercury species determination by HPLC-ICP-MS
Analyses were performed in time resolved analysis mode. The experimental conditions are given in Table I. Each sample was analyzed four times to enable statistical evaluation of the samples (n = 3 × 4).
EPA Method 6800 for SIDMS analysis
To perform SIDMS analysis, the crude oil sample was weighed into microwave vessels. Before extraction, analytically appropriate amounts of 199 Hg2+, CH3200 Hg+, and C2H5201 Hg+ were added to each sample in the microwave vessel to result in a ratio of measured isotope that was close to the optimum isotope ratio for each species of interest. Optimization of spiking factors was done by calculating isotopic enrichments and each natural isotope combination for each isotope (15). The optimum amount of isotope spike depends upon the relative levels of inorganic mercury, monomethylmercury, and monoethylmercury present in the sample, the enrichment purity of the reagent isotope and the natural ratio of that corresponding isotope as they occur in nature. The optimum spike level is achieved by knowing the aforementioned values and, for a particular standard, by obtaining the certified value of each species from the certificate of analysis in the AIT spike kit, or in an unknown with prior workup of unspiked sample to estimate the approximate concentration of each species present in the sample. To obtain accurate and precise measurements with IDMS and SIDMS analyses, the optimum amount of isotopic analogues of inorganic mercury, monomethylmercury, and monoethylmercury were calculated to spread over one order of magnitude (that is, the sample to spike ratio should be in between 0.01 and 1). During this study, an optimum isotope ratio of 0.1:1 (sample:spike) was used. The samples were allowed to equilibrate for 1 h and were then extracted according to EPA Method 3200. Extracts were analyzed using HPLC–ICP-MS. The experimental conditions are given in Table I.
Peak areas were used to calculate isotope ratios 199 Hg/202 Hg and 200 Hg/202 Hg, from which the Hg2+, CH3Hg+, and C2H5201 Hg+ concentrations in crude oil were calculated. All isotope ratios were corrected for the detector dead time, and a natural abundance standard solution of Hg2+, CH3Hg+, and C2H5201 Hg+ was measured periodically between samples to calculate the mass bias correction factor. Further, this triple spike approach allowed tracking of any artifact methylation–demethylation or ethylation–deethylation reactions that occurred during the sample preparation and/or analysis process. The calculations were carried out using the software provided in the enriched isotopic species kit by AIT, as specified in EPA Method 6800. The SIDMS protocol utilizes direct mathematical algorithmic solutions for isotope ratio calculations and quantitative determinations instead of conventional calibration curves. These direct solutions, provided by the 3Hg-SPC software (Applied Isotope Technologies), were used as specified in EPA Method 6800 for all species calculations.
Results and Discussion
Determination of total mercury with EPA Method 3052 and 6800 (IDMS)
While species of mercury in the crude oil samples were the primary analytical targets, total mercury concentrations are also critical so that mass balance can be calculated by adding all the species components. A different sample preparation from the speciated extraction method was required to determine the total mercury content in the crude oil sample that was digested according to EPA Method 3052. The digest was then analyzed with ICP-MS in the spectrum mode. The concentration of total mercury was then determined from the IDMS calculation software for total mercury. The determined results are reported in Table II. The measured total mercury concentrations were 414.1 ± 24.7 μg/kg (No. 871833) and 437.8 ± 25.4 μg/kg (No. 870987) for two samples of crude oil respectively. These became the reference values for mass balance of species components in the samples.



Table II: Total mercury concentration in crude oil samples by ID-ICP-MS analysis
Determination of soluble mercury species by EPA Method 3200 and 6800 (SIDMS)
The application of SIDMS depends upon some fundamental operations: enriched stable speciated isotopic spike preparation and calibration or purchase of the enriched stable isotopic spike analogue; sample collection and sample spiking; sample species and spike species equilibration; sample extraction; species separation; isotope ratio measurements of each speciated component; and determination of species concentrations and mathematical deconvolution of species transformations. The last step of the method involves a direct mathematical solution that eliminates the need to use traditional calibration curves for concentration calculations. Species transformations that would have produced biases with other methods are calculated and corrected for in the SIDMS mathematical protocol specified in EPA Method 6800.
To perform SIDMS analysis, a different set of subsamples was processed using the same crude oil samples with EPA Method 3200. A known amount of crude oil sample was triple spiked with known amounts of isotopically enriched stable inorganic mercury (199 Hg2+), monomethylmercury (CH3200 Hg+) and monoethylmercury (C2H5201 Hg+) in such a way that the desired isotope ratios 199 Hg/202 Hg, 200 Hg/202 Hg, and 201 Hg/202 Hg became close to the optimum required isotope ratio (0.1:1, sample:spike). After equilibration with the sample species, the samples were extracted using EPA Method 3200, and the extracts were analyzed with HPLC–ICP-MS. The deadtime and mass bias corrected isotope ratios for 199 Hg/202 Hg, 202 Hg/202 Hg, and 201 Hg/200 Hg were calculated for inorganic mercury, monomethylmercury, and monoethylmercury in each of the sample replicates. The SIDMS calculations were performed to determine the concentrations of inorganic mercury, monomethylmercury, and monoethylmercury, and to deconvolute the interspecies transformations using the software described earlier. In this software, the concentration and correction calculations are conveniently done because the isotopic values and ratios are keyed to the specific enriched isotopic reagent batch supplied in the speciation kit. A detailed description of data processing and application of SIDMS software algorithms has been reported elsewhere (16,17).
The final concentrations of inorganic mercury, monomethylmercury, and monoethylmercury in crude oils and the percent of interspecies transformation during extraction are summarized in Table III. Table III demonstrates that the concentration of inorganic mercury ranged from 17.3 ± 3.6 μg/kg to 23.6 ±3.7 μg/kg, monomethylmercury ranged from 0.5 ± 0.1 to 0.8 ± 0.1 μg/kg, and monoethylmercury ranged from 7.4 ± 2.2 to 10.7 ± 1.1 μg/kg.



Table III: SID-ICP-MS analysis of three soluble mercury species from crude oil after EPA Method 3200 extraction
The percentage of mercury species transformations obtained using the studied extraction methods also is shown in Table III. It is observed that the degree of deethylation to inorganic mercury was severe for both crude oil samples and was in the range of 55.1% and 86.9%, whereas the demethylation to inorganic mercury was less than 7% in both samples and was in the range of 2.9% and 6.2%. The degree of methylation from inorganic mercury and monoethylmercury was less than 10% in both crude oil samples and was in the range of 6.1 % and 9.6%. The degree of ethylation from both inorganic mercury and monomethylmercury was less than 3% and was in the range of 0.8 % and 2.1%. Because of the relatively similar ratio of Hg2+ to C2H5Hg+ in crude oil samples, high deethylation would have caused a significant error if external calibration protocol was used for this study.
Determination of nonsoluble mercury fractions from EPA Method 3200 residues by EPA Method 3052 and 6800 (IDMS)
The oil residues left over after EPA Method 3200 extraction of soluble mercury fractions were respiked with known amount of isotopically enriched inorganic mercury (199 Hg+) and 9 mL of concentrated HNO3, and 1 mL of 30% H2O2 was added in microwave vessels. The samples were then digested according to EPA Method 3052 and the digests were analyzed with ICP-MS in the spectrum mode. The concentration of the total nonsoluble mercury fraction was then calculated by using the software. The results for the nonextractable mercury fraction are reported in Table IV. It is observed from Table IV that the nonextractable mercury fractions in the crude oil samples were 342.5 ± 46.8 μg/kg (No. 871833) and 323.3 ± 30.7 μg/kg (No. 870987).


Table IV: ID-ICP-MS analysis of nonsoluble mercury in crude oil residue after EPA Method 3200 extraction
To perform the mass balance, the concentration of all mercury species from the soluble mercury and nonsoluble mercury were added arithmetically. The total mercury concentrations were found to be 370.2 ± 47.0 μg/kg (No. 871833) and 354.8 ± 31.0 μg/kg (No. 870987). It is observed that the mass balance value and that of the total mercury value for the No. 871833 sample were statistically indistinguishable, whereas the mass balance value for the No. 870987 was 7% less than that of the total mercury value obtained from IDMS analysis.
Conclusions
The determination of total mercury and different mercury species in soluble mercury fractions as well as in the nonsoluble mercury fraction was performed on two crude oil samples using both IDMS and SIDMS protocols described in EPA Method 6800. Results for total mercury and mass balance from one crude oil sample agreed statistically at their 95% CL, whereas that for the sample was 7% different from total mercury value determined by IDMS technique. In both samples, the amounts of soluble mercury species were approximately 7% of the total mercury value. The interconversion from monoethylmercury to inorganic mercury was high in both samples and ranged from 55% to 87%, whereas the methylation was less than 10% and ethylation was less than 3% in both samples. The alkyl mercury species are reactive and interconvert during analyses steps and require correction to achieve accurate results. The remaining nonextractable mercury species are the subject of further protocol developments. We have shown here that significant interdependent concentrations of three alkyl mercury species in the natural crude oil samples can be determined with relatively high accuracy after optimization and application of a combination of three environmental measurement protocols.
Acknowledgments
The authors wish to thank and acknowledge the collaboration of our colleagues for samples and interaction in these studies. We thank Milestone Inc, Metrohm Peak, Agilent Technologies, Applied Isotope Technologies, Inc., and Duquesne University for instrumental and material support. Portions of this research are patented or have patents pending.
G.M. Mizanur Rahman, Timothy M. Fahrenholz, and H. M. Skip Kingston are with Duquesne University, Pittsburgh, Pennsylvania, Matt Pamuku is with Applied Isotope Technologies, Inc., Sunnyvale, California, and J. David Hwang and Lyman A. Young are with Chevron Energy Technology Company, Richmond, California.
References
(1) M.A. McDowell, C.F. Dillon, J. Osterloh, P.M. Bolger, E. Pellizzari, R. Fernando, R. Montes de Oca, S.E. Schober, T. Sinks, R.L. Jones, and K.R. Mahaffey, Environ. Health Perspect. 112(11), 1165–1171 (2004).
(2) G.P. Brandao, R.C. de Campos, and A.S. Luna, Spectrochim. Acta, Part B 60, 625 (2005).
(3) S.M. Wilhelm, L. Liang, D. Cussen, and D.A. Kirchgessner, Environ. Sci. Technol. 41(13), 4509 (2007).
(4) T.Y. Yan, Ind. Eng. Chem. Res. 35, 3697 (1996).
(7) B. Bouyssiere, F. Baco, L. Savary, and R. Lobinski, J. Chromatogr. 976, 431 (2002).
(5) N.S. Bloom, Fresenius, J. Anal. Chem. 366, 438 (2000).
(6) W.R. Kelly, S.E. Long, and J.L. Mann, Anal. Bioanal. Chem. 376, 753 (2003).
(8) L. Liang, M. Horvat, and N.S. Bloom, Talanta 41(3), 371 (1994).
(9) C. Schickling and J.A. Broekaert, Appl. Organomet. Chem. 9, 29 (1995).
(10) G. Westoo, Acta Chem. Scand. 20, 2131 (1966).
(11) J.P. Snell, W. Frech, and Y. Thomassen, Analyst 121, 1055 (1996).
(12) L.H. Reyes, G.M.M. Rahman, T. Fahrenholz, and H.M. Kingston, Anal. Bioanal. Chem. 390, 2123 (2008).
(13) US EPA Method 3052 in Test Methods for Evaluating Solid Waste, Physical/Chemical Methods SW 846, U.S. Government Printing Office, Washington, DC, 1996.
(14) U.S. EPA Method 6800 in Test Methods for Evaluating Solid Waste, Physical/Chemical Methods SW 846, Update IVA, US Government Printing Office, Washington, DC, 2008.
(15) J.I. Garcia-Alonso, Anal. Chim. Acta 312, 57–78 (1995).
(16) G.M.M. Rahman and H.M.S. Kingston, Anal. Chem. 76, 3548–3555 (2004).
(17) G.M.M. Rahman, Ph.D. Dissertation, Duquesne University, 2004.


















LIBS Illuminates the Hidden Health Risks of Indoor Welding and Soldering
April 23rd 2025A new dual-spectroscopy approach reveals real-time pollution threats in indoor workspaces. Chinese researchers have pioneered the use of laser-induced breakdown spectroscopy (LIBS) and aerosol mass spectrometry to uncover and monitor harmful heavy metal and dust emissions from soldering and welding in real-time. These complementary tools offer a fast, accurate means to evaluate air quality threats in industrial and indoor environments—where people spend most of their time.


NIR Spectroscopy Explored as Sustainable Approach to Detecting Bovine Mastitis
April 23rd 2025A new study published in Applied Food Research demonstrates that near-infrared spectroscopy (NIRS) can effectively detect subclinical bovine mastitis in milk, offering a fast, non-invasive method to guide targeted antibiotic treatment and support sustainable dairy practices.

Smarter Sensors, Cleaner Earth Using AI and IoT for Pollution Monitoring
April 22nd 2025A global research team has detailed how smart sensors, artificial intelligence (AI), machine learning, and Internet of Things (IoT) technologies are transforming the detection and management of environmental pollutants. Their comprehensive review highlights how spectroscopy and sensor networks are now key tools in real-time pollution tracking.


