The State of the Art of Flow-Through Solid-Phase Spectrometry
Special Issues
Flow-through SPS lowers reagent and sample consumption and decreases waste generation.
Sample pretreatment is one of the bottlenecks in analytical chemistry, especially when dealing with complex matrices like environmental samples. When performed in a batch mode, sample handling methods are tedious and time consuming. Therefore, the hyphenation of these methods with flow-injection techniques yields many advantages. The possibility of automation not only increases the determination rate, but also decreases sample and reagent consumption. As a consequence, analyte separation, enrichment, and elimination of sample matrix becomes possible with an increase in selectivity and sensitivity. This is a significant contribution for the analysis of environmental samples because the analyte is usually present at trace levels in a complex matrix. In this scenario, the state of the art of solid-phase spectrometry (SPS) with a focus on the lab-on-valve (LOV) platform is discussed. LOV facilitates the manipulation of bead suspension for SPS with lower reagents consumption and waste production.
When analyzing environmental samples such as water, soil, and plants, some major challenges may be found. For example, when dealing with dynamic systems such as estuarine waters, spatial and temporal variability may be encountered. For this reason, the analyte concentration may range from low to trace levels. Salinity in estuarine systems may be a good example, because it presents both spatial variability (proximity to the sea) and temporal variability (tides). Solid environmental samples, such as soil and plants, are another example where difficulties may be found because some type of extraction is needed to isolate and separate the analyte from its matrix. Because of these challenges, a sample pretreatment is often necessary before identifying or quantifying the analyte of interest to increase the method’s selectivity and sensitivity. Different separation techniques such as liquid–solid extraction, liquid–liquid extraction (LLE), and gas chromatography (GC) and liquid chromatography (LC) are available to overcome these issues. In this manner, analyte extraction and enrichment can be performed along with the removal of sample matrix interferences. However, when performed in a batch mode, these sample pretreatment methods are very tedious and time-consuming. Furthermore, high amounts of organic solvents are usually necessary, especially for solvent extraction methods, which can cause health and environmental problems because of their high volatility and release into the environment (1).
In this context, the coupling of separation techniques with flow-injection methods not only allows the automation of the entire sample preparation process, but also achieves a reduction in reagent and sample consumption. Also, an increase in the sensitivity of the method can be obtained together with an increase in throughput (1,2). By incorporating external devices, such as gas diffusion or dialysis units, or resin packed columns, in the flow manifolds, the analyte of interest can be collected, enriched, and separated from its matrix before detection in a miniaturized fashion.
Different flow techniques can be used according to their suitability for the intended determination. Flow-injection analysis (FIA) was first described by Ruzicka and Hansen in 1975 (3) where the concept of complete reaction and physical equilibrium was discarded. The sample is injected in a continuous flow of reagent and the mixture is performed as the sample is propelled downstream to the detector. To overcome some of the FIA limitations, a second generation was proposed as an evolution to this technique. The main principle of sequential-injection analysis (SIA), the so-called second generation of flow injection analysis, is the programmable flow where the mixing occurs by reversing the flow of sample and reagents (4). With this principle, SIA allows an even lower consumption of reagents and effluent production. The third generation of flow injection analysis, called sequential injection lab-on-valve (SI-LOV), has the main characteristics of SIA (5). However, this technique incorporates the detection system in the selection valve, which allows a working volume in the microliter range. Additionally, this technique allows handling solid materials within the manifold conduits in a relatively simple way. This feature opens new perspectives for performing several processes on the sorbent surface, such as analyte enrichment, immobilization of reagents, and derivatization reactions. If the solid material is sufficiently transparent, even the spectrometric measurement itself can be made directly on the solid material-that is, solid-phase spectrometry (SPS). This approach is quite a breakthrough for samples with complex matrices such as environmental samples. SPS provides the ability to minimize possible physical interferences (those caused by sample intrinsic color or turbidity) and chemical interferences. Also, as already mentioned, the analyte is usually present at trace levels, so this technique allows the enrichment as well as sample cleanup (for example, desalting). Additionally, a sensitivity enhancement can be achieved because there is no need for a previous elution before measurement. This feature is explored in this article.
Hence, the coupling of FIA, SIA, and SI-LOV with on-line sample pretreatment procedures offers different advantages when compared to performing these procedures in a batch mode. Each technique presents unique characteristics that can contribute to the automation of the sample pretreatment procedure. FIA requires a simpler manifold, and because of its continuous flow, a higher throughput can be achieved. SIA and SI-LOV, in contrast to FIA, require a more sophisticated manifold; however, since they are based on a programmable flow, even lower sample and reagent consumption can be achieved. Also, since a selection valve is used instead of an injection valve, different reagents and devices can be coupled and therefore multiparametric determinations can be performed with the same manifold. In the meantime, other flow techniques have been described such as multicommuted flow injection analysis (MCFIA), multisyringe flow injection analysis (MSFIA), and multi-pumping flow analysis (MPFA) (6–8). Since they are all based on a unidirectional flow like flow-injection analysis-and to facilitate the reading of this article-all of these techniques were included in the FIA group. This article discusses the state of the art of flow-through SPS. Furthermore, it provides a detailed review on the use of SI-LOV for SPS.
On-Line Sample Pretreatment
The advantages of coupling flow techniques with on-line sample pretreatment procedures are diverse. In this section, we focus on extraction and preconcentration as sample pretreatment techniques. In this context, liquid–solid extraction or solid-phase extraction (SPE), LLE, GC, LC, and membrane-based techniques such as gas diffusion, dialysis, and pervaporation, are the techniques included in this section for discussion. SPE, LLE, GC, and LC are techniques used for separation of the analyte from possible sample matrix interferences. SPE and LLE moreover can be used to enrich the analyte in a solid or liquid phase, respectively. Membrane-based techniques can also be used for analyte separation from sample matrix as the species are transferred through a membrane from a donor to an acceptor solution. The difference between dialysis and gas diffusion is the membrane material. These techniques can also be performed with the purpose of dilution and microextraction.
A search on ISI Web of Knowledge-Web of Science (Figure 1) was made for the existing publications (between the years 2000 and 2015) that couple sample pretreatment methods with flow techniques.
As shown in Figure 1, SPE is the first choice for on-line sample pretreatment in all flow techniques. In comparison, there have been few papers describing the hyphenation of LLE with flow techniques. This method still requires the use of organic solvents, although in lower volumes when compared to batch LLE, which may be the cause for it being used less when compared to other pretreatment methods.


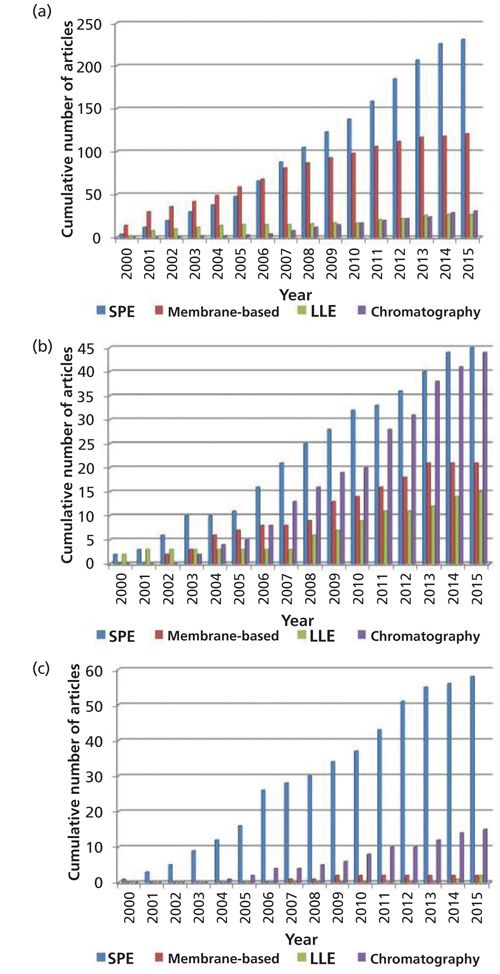
The coupling of membrane-based methods to flow techniques constitutes an excellent tool for the monitoring of dynamic systems. These on-line pre-treatment methods have been reasonably used throughout the years, with more application in the FIA and SIA methods. The yields of these membrane-based methods can be optimized and adapted to the intended application (for example, separation, enrichment, or dilution). However, when coupled to flow injection techniques, the obtained yields are usually quite low as flow techniques frequently present a short time available for analyte transfer. This may be one of the reasons for the decrease in published works that use this on-line sample pretreatment method.
Chromatography has been well explored using SIA methods. In fact, a significant increase in the published papers where chromatography is hyphenated to SIA is shown in Figure 1b. This increase can be explained by the recent development of monolithic columns (9–11) that, because of their porosity, allow efficient separations at lower pressure. Although this was a good contribution, chromatography usually requires a step of analyte enrichment before separation, which in turn makes the method more complex.
SPE coupled to flow techniques has been well explored throughout the years (1,12,13). Indeed, this method has resulted in the improvement of simplicity and ease of automation when compared to the batch mode. By introducing a packed resin to the flow manifold, analyte separation and enrichment can be achieved in a few steps. In doing so, the method’s throughput, sensitivity, and selectivity may be increased.
Flow-Through SPS
The performance of on-line SPE was a great advance in the automation of sample pretreatment. In this context, SPS was described for the first time by Yoshimura and colleagues (14). Both techniques are based on analyte retention on a solid support. In SPE, the analyte must be eluted from the solid support toward the detector, but in SPS the beads are trapped in a flow cell where the signal is measured at the surface of the beads (optosensing). When compared to the traditional SPE technique, SPS does not require the step of analyte elution from the solid support where dilution and partial loss of the preconcentration capabilities may occur. In fact, SPS not only allows analyte retention and matrix interference elimination, it also reduces intrinsic sample and bead absorption by resetting the absorbance baseline value after propelling the sample through the packed beads. Therefore, this technique exhibits high sensitivity and selectivity provided because of the in situ preconcentration and detection of analytes on the solid sensing support.
SPS has been mainly performed using FIA, as shown in Figure 2, probably because of the simplicity of the manifold.


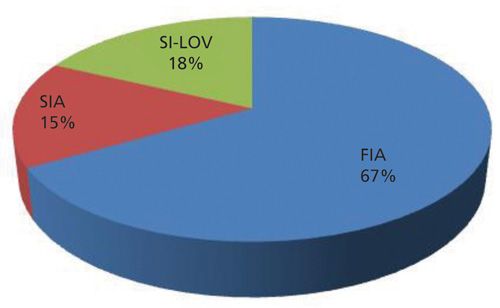
The great advantage of performing flow-through SPS when compared to batch SPS, besides consuming less reagents and producing less waste, is the possibility of bead reutilization, as they are regenerated after each determination. Therefore, SPS is a good contribution to the green analytical chemistry concept because there is a reduction in sample, reagent, and solid support consumption and effluent production by downscaling the analytical system.
Different approaches can be used for SPS (15). In the first approach, the analyte is primarily retained on the beads and then the chromogenic reagent is added. This procedure is mainly applied when the reagent has poor selectivity for the analyte. For this reason, possible matrix interferences must be removed so the analyte is the only species available to react with the chromogenic reagent. In the second approach, the reagent is first retained on the beads, functionalizing them, and then the sample is added. This procedure is recommended when the color reaction is highly selective for the analyte and the product formed can be sorbed on the solid support. Another approach performed is the measurement of the analyte intrinsic absorbance or fluorescence without the need of using a chromogenic reagent.
Detection Methods
To perform flow-through SPS, only a few detection methods are available because of the need to measure the signal at the surface of the beads. The major problem associated with this approach is that, due to the packed sorbent in the flow cell, a large background signal is already measured before the determination. As there is a large background attenuance of the solid phase, a relatively small absorbance of the colored species adsorbed on the solid phase is measured, which can decrease the sensitivity of the method.
Consequently, the detection methods described in the literature used for flow-through SPS are ultraviolet–visible (UV–vis) molecular absorption, fluorescence, chemiluminescence, phosphorescence, and reflectometry (Figure 3), because these allow the measurement of a signal at the surface of a solid support (16).


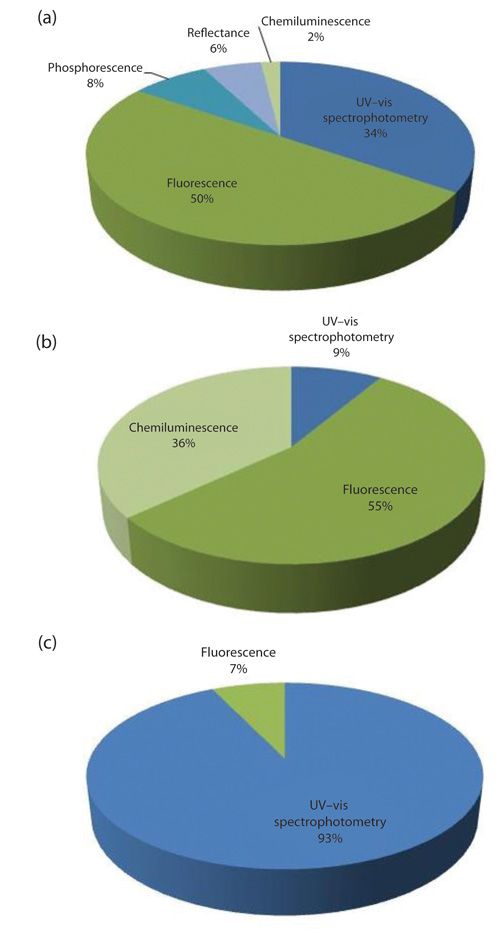
According to Figures 3a and 3b, fluorescence is the most common detection method in FIA and SIA techniques for flow-through SPS. Fluorescence is more sensitive and selective than UV–vis spectrophotometry; it is subject to less influence from the solid material background signal and interferences from the sample matrix.
However, for SI-LOV, UV–vis is clearly the most common detection device used. In fact, the flow cell of the SI-LOV can be configured for UV–vis and fluorescence measurements by means of optical fibers. However, fluorescence is not as sensible as UV–vis molecular absorption when performed in this platform. Usually, higher flow paths (1 or 1.5 cm) are advised for fluorescence measurements and the flow path of SI-LOV can only be 1 cm maximum, if no additional device, such as a Garth cell (17), is used.
Lab-on-Valve Platform for SPS
The development of SI-LOV was a big step toward miniaturization and automation of chemical analysis. Its new design, which integrates the flow cell on top of the multiposition valve, made the reduction of sample and reagent volumes and effluent production (5) possible. Moreover, the geometry of the channels in the multiposition valve of the SI-LOV allows the manipulation of beads that can be trapped in different places in the valve, an approach called bead injection (BI). The use of beads and their entrapment in the selection valve allows the performance of SPE or chromatography without the need for coupling external devices such as columns. Furthermore, if the beads are entrapped in the flow cell of the multiposition valve, the analyte can be retained and determined at the surface of the beads (SPS). When compared to FI-SPS and SIA-SPS, SI-LOV-SPS offers the possibility to renew the adsorbents not only by chemical regeneration (elution), but also by physical regeneration, where the beads are discarded and a new sensor is prepared in the flow cell after each analytical cycle. By doing so, no elution step is necessary to clean the sensor and thus, no analyte or interfering species accumulation occurs. Therefore, the lifetime of the sensor is not a limitation of the method. Bead injection in an LOV platform for SPS simplified the on-line sample pretreatment procedure because column preparation, analyte retention, enrichment, detection, and elution-washing can be performed automatically by computer control (18).
Absorbance and fluorescence measurements can be carried out in the flow cell that is integrated within an LOV module by means of optical fibers. The distance between the optical fiber ends defines the optical pathlength, which can be varied from 1 mm (Figure 4b) to 10 mm (Figure 4c). Fluorescence measurements (Figure 4a) are carried with optical fibers assembled at a 90° angle.


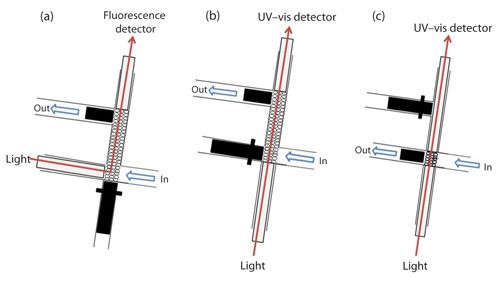
The use of a higher flow path may increase the sensitivity of the method because a higher mass of resin can be packed in the flow cell. On the other hand, a higher amount of sensor in the flow cell may cause higher background signal and therefore low analytical signal decreasing the sensitivity of the method.
A review of all the works describing the use of SI-LOV for SPS is presented in Table I.


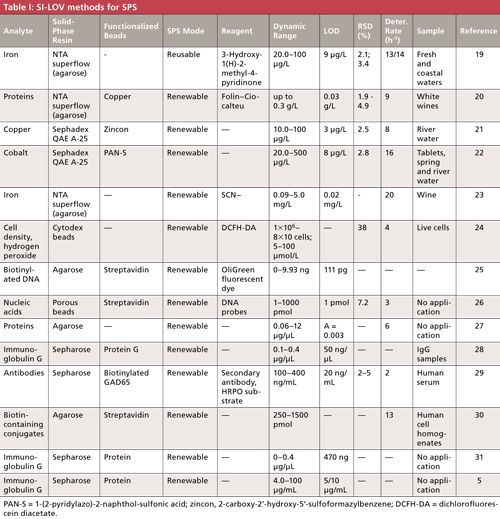
Almost all works use functionalized beads, where the reagent has been previously retained on the surface of the beads. This approach can increase throughput, because there is no need to aspirate the chromogenic reagent. However, the chromogenic reagent must be selective towards the analyte; otherwise, there may be some possible interferences from the sample matrix. In fact, some researchers not only functionalize the beads, but also use a second reagent that is propelled after the analyte is retained. By doing so, even higher selectivity is obtained.
Almost all the described works use the renewable approach, which means that the sorbents are not reused but renewed after each determination. As previously discussed, this method is not limited by the sensor’s lifetime and no accumulation, either of the analyte or of the interfering species, is observed. When comparing the methods that use the reusable or the renewable approach, no significant difference in the throughput is observed. Higher determination rates would be expected for the reusable approach as the sensor is not built after each analytical cycle. However, the need to elute and wash the beads in the reusable approach, also requires some time, which can decrease the determination rate.
As shown in Table I, the SI-LOV-SPS methods were applied to samples with complex matrices such as human serum, wine, and fresh and coastal waters. In fact, the more recent works (19,21,22) were applied to water samples, showing the increased need for new and more sensitive methods that can be applied to these complex samples. The presented works describe the efficient removal of potential sample matrix interferences and the methods were successfully applied to the determination of the analyte of interest at trace levels. In fact, limits of detection at the microgram-per-liter level were achieved, demonstrating the increased sensitivity and selectivity obtained when performing flow-through SPS.
The chosen material for the solid support was agarose because it is easily coated with different molecules to modify its affinity according to the intended determination. This is also a good choice for bead injection in an LOV platform as explained in the following section.
Adsorbent Characteristics
For the efficient retention and enrichment of analytes, and for the efficient removal of possible interfering matrix substances, a suitable sorbent must be selected. The interaction of the analyte with the solid support is of extreme importance, but the adsorbent itself must fulfill some requirements so it can be used as an optosensor in flow-through SPS. The adsorbent characteristics are more important when performing SPS in a bead injection mode. When the sensor is manually placed in the flow cell, there is only the need for optical transparency to prevent high signal background. In the bead injection mode, as the optosensor is built by aspirating the beads through the manifold tubing, certain size and material requirements are necessary to prevent clogging and scratching of, for example, the LOV channels. According to Ruzicka (17), the particles must be spherical with a size in the range of 20 to 150 µm. The bead size must be homogeneous to ensure reproducibility in the SPS method when bead injection is performed in a renewable approach. Soft polymer beads are preferable, as rigid beads may scratch the selection valve. Sephadex and sepharose beads are therefore a good choice for bead injection applications because they fulfill the mentioned criteria by being globe-shaped and of regular size (13,32).
Conclusions
Automation of sample pretreatment techniques is of great interest because they are tedious and time consuming and consume large amounts of toxic reagents. The hyphenation of flow techniques with sample pretreatment techniques was a breakthrough in analytical chemistry because the automation of these tedious methods was possible. Flow-through SPS has been a significant contribution to this field. This technique allows the analyte separation and enrichment by removing possible sample matrix interferences with detection on the surface of the sensor. Therefore, higher sensitivity and selectivity is achieved when comparing this technique with other on-line sample pretreatment techniques. SI-LOV-SPS was an even bigger advance as sensor preparation, analyte retention, enrichment, detection, and washing can be performed automatically in few steps by computer control.
Nowadays, the concept of green chemistry receives greater attention to prevent environmental pollution by chemical activities. The main aim is to minimize or eliminate reagent consumption and waste production if possible by automation and miniaturization of the analytical systems. Flow-through SPS is a good contribution in this field because it allows a lower reagent and sample consumption with a decrease in waste generation.
In spite of presenting several benefits in on-line sample pretreatment, flow-through SPS also has some disadvantages. Since the sensor is built in the flow cell, high background signal can be experienced that may decrease the sensitivity of the method. Also, a gain in sensitivity by increasing the sensor length is difficult, especially when using SI-LOV because the flow path is limited to a maximum of 1 cm.
Acknowledgments
I.C. Santos thanks Fundação para a Ciência e a Tecnologia (FCT, Portugal) and Fundo Social Europeu (FSE) through the program POPH-QREN for the grant SFRH/BD/76012/2011. This work was supported by National Funds through FCT, through projects PTDC/AAG-MAA/3978/2012, and PEst-OE/EQB/LA0016/2013.
References
(1) S. Motomizu and T. Sakai, in Comprehensive Analytical Chemistry: Advances in Flow Injection Analysis and Related Techniques, 1st Ed. (Elsevier, The Netherlands, 2008), pp. 159–199.
(2) M.D.L. de Castro, in Comprehensive Analytical Chemistry: Advances in Flow Injection Analysis and Related Techniques, 1st Ed. (Elsevier, The Netherlands, 2008), pp. 204–232.
(3) J. Ruzicka and E.H. Hansen, Anal. Chim.Acta78, 145–147 (1975).
(4) J. Ruzicka and G.D. Marshall, Anal. Chim. Acta 237, 329–343 (1990).
(5) J. Ruzicka, Analyst 1053–1060 (2000).
(6) B.F. Reis, M.F. Gine, E.A.G. Zagatto, J.L.F.C. Lima, and R.A. Lapa, Anal. Chim. Acta 293, 129–138 (1994).
(7) V. Cerdà, J.M. Estela, R. Forteza, A. Cladera, E. Becerra, P. Altimira, and P. Sitjar, Talanta 50, 695–705 (1999).
(8) R.A.S. Lapa, J.L.F.C. Lima, B.F. Reis, J.L.M. Santos, and E.A.G. Zagatto, Anal. Chim. Acta466, 125–132 (2002).
(9) F. Svec and C.G. Huber, Anal. Chem.78, 2100–2107 (2006).
(10) D. Šatínský, P. Solich, P. Chocholouš, and R. KarlíÄek, Anal. Chim. Acta 499, 205-214 (2003).
(11) M. Fernández, H.M. González-San Miguel, J.M. Estela, and V. Cerdà, TrAC-Trends Anal. Chem.28, 336–346 (2009).
(12) M. Miró and E.H. Hansen, TrAC-Trends Anal. Chem.25, 267–281 (2006).
(13) M. Miró, S.K. Hartwell, J. Jakmunee, K. Grudpan, and E.H. Hansen, TrAC-Trends Anal. Chem.27, 749–761 (2008).
(14) K. Yoshimura, H. Waki, and S. Ohashi, Talanta23, 449–454 (1976).
(15) A. Molina-Díaz, J.F. García-Reyes, and B. Gilbert-López, TrAC-Trends Anal. Chem.29, 654–666 (2010).
(16) S. Matsuoka and K. Yoshimura, Anal. Chim. Acta664, 1–18 (2010).
(17) Tutorial on Flow Based microAnalytical Techinques, J. Ruzicka, Ed. (CD, http://www.flowinjection.com, USA, 4th Ed., 2009).
(18) S.S.M.P. Vidigal, I.V. Tóth, and A.O.S.S. Rangel, Anal. Methods5, 585–597 (2013).
(19) R. Suárez, R.B.R. Mesquita, M. Rangel, V. Cerdà, and A.O.S.S. Rangel, Talanta 133, 15–20 (2015).
(20) S.S.M.P. Vidigal, I.V. Tóth, and A.O.S.S. Rangel, Talanta96, 102–106 (2012).
(21) Y.-L. Yu, Y. Jiang, and R.-H. He, Talanta88, 352–357 (2012).
(22) Y.-L. Yu, Y. Jiang, S. Dou, and J.-H. Wang, Anal. Methods4, 1718–1724 (2012).
(23) S.S.M.P. Vidigal, I.V. Tóth, and A.O.S.S. Rangel, Talanta84, 1298–1303 (2011).
(24) I. Lähdesmäki, P. Chocholous, A.D. Carroll, J. Anderson, P.S. Rabinovitch, and J. Ruzicka, Analyst134, 1498–1504 (2009).
(25) M. Decuir, I. Lähdesmäki, A.D. Carroll, and J. Ruzicka, Analyst132, 818–822 (2007).
(26) K.A. Edwards and A.J. Baeumner, Anal. Chem.78, 1958–1966 (2006).
(27) J. Ruzicka, A.D. Carroll, and I. Lähdesmäki, Analyst131, 799–808 (2006).
(28) Y. Gutzman, A.D. Carroll, and J. Ruzicka, Analyst 131, 809–815 (2006).
(29) A.D. Carroll, L. Scampavia, D. Luo, Å. Lernmark, and J. Ruzicka, Analyst128, 1157–1162 (2003).
(30) Y. Ogata, L. Scampavia, J. Ruzicka, C.R. Scott, M.H. Gelb, and F. TureÄek, Anal. Chem. 74, 4702–4708 (2002).
(31) A.D. Carroll, L. Scampavia, and J. Ruzicka, Analyst127, 1228–1232 (2002).
(32) H.M. Oliveira, M. Miró, M.A. Segundo, and J.L.F.C. Lima, Talanta84, 846–852 (2011).



















New Study Reveals Insights into Phenol’s Behavior in Ice
April 16th 2025A new study published in Spectrochimica Acta Part A by Dominik Heger and colleagues at Masaryk University reveals that phenol's photophysical properties change significantly when frozen, potentially enabling its breakdown by sunlight in icy environments.




