Those Ions Know What They Are Doing: Part II
Here, we expand on some of the central tenets of models introduced in part I and describe the transition state invoked in unimolecular dissociation reactions.



In part I of this column series, we dispelled the notion of a system temperature as a useful metric in assessing the rate of ion dissociation reactions occurring in a mass spectrometer. We reviewed the energetics of ions formed by chemical ionization (perhaps the simplest example to follow) and tracked the differing degrees of dissociation as a function of the internal energy in the protonated molecule. In part II of this series, we expand on some of the central tenets of models introduced in part I and describe the transition state invoked in unimolecular dissociation reactions. The ultimate test of the validity of the theoretical and calculational models remains a comparison of predicted and observed mass spectra. With newer ionization methods, spectra recorded under various experimental conditions are examined for insights into the energetics of the ionization process. Experimental results always inform the assessment and development of theory. In a simplistic aphorism, theory guides and experiment decides.
In an electron ionization (EI) source, the pressure of the sample molecules is low. The neutral sample molecules are few and far between, and at a density that is similar to the background pressure of the source itself. The ionization process occurs as the result of the interaction between an energetic electron and a single sample molecule; a good EI source confines the ionization within as small a volume as possible that is consistent with the constraints of space charge. The ionization process produces a positive ion with a certain amount of internal energy because of the ejection of an electron. The dissociation of this positive ion occurs in isolation (there is no collision with other species in the source at low pressures), and is to an extent governed by that amount and distribution of internal energy within the ion. Figure 1 illustrates a central concept in statistical rate theory models that describe the unimolecular dissociation reactions — the activated complex or transition state as a species on the plot of ion internal energy (y-axis) and reaction coordinate (x-axis). A reaction occurs as the system moves to the right along the reaction coordinate. A dissociation reaction occurs if the ion has sufficient energy to exceed the activation energy and achieve the structure of the activated complex.


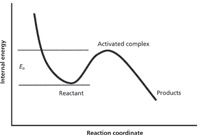
Figure 1: Schematic potential energy diagram for an ion following a specific dissociation reaction. Progress in the reaction corresponds to movement along the x-axis of the reaction coordinate. If the ion internal energy E exceeds the activation energy E0, the structure of the activated complex may be attained. In the activated complex, bonds are stretched and broken as the ion dissociates to form the a fragment ion. This diagram is a slice through a more complex three-dimensional potential energy surface that may have several maxima and minima, representing several different reaction paths. A consecutive reaction could appear in this diagram with a second activation energy hump and a different final end product energy.
So, just what is the activated complex? The structure of the activated complex is achieved long after the initial ionization has taken place, and long after any ion internal energy has been distributed throughout the ion. The energy of the complex is not only the energy required to achieve it, but also any added energy that it contains, which is dependent on the ionization process. We can assume that the activated complex has a structure different from that of the original neutral molecule. In the activated complex, bonds are stretched and bent as the ion "explores" its potential for dissociation. The activated complexes can be described as "loose," a structure in which all rotations are possible. If the activated complex is "tight," some rotations are not allowed, at least in theory. These mathematical abstractions are described by casual terms to allow a connection to a specific structure and provide a rational path in cleavages and rearrangements to the final ionic products. In statistical rate theories, the activated complex is described as a collection of oscillators, with specific enumerated vibrational and rotational states. A neutral molecule is analogously described, but of course, the exact energy levels of the states differ. The rate of the dissociation reaction of the ion depends on the values of the activation energy, the total internal energy, and the number of states. Additionally, the rate of a particular reaction depends on whether there are other discrete reactions with similar activation energies and state densities.
Fundamental Assumptions of Statistical Rate Theories
Let's revisit the fundamental assumptions of statistical rate theories introduced in the last column:
- Ion dissociation reactions occur much more slowly than the initial ionization process.
In electron ionization, the removal of an electron from a neutral molecule to form the odd-electron molecular ion occurs in about 10–16 s. This vertical Franck-Condon transition results in ionization without any changes in the atom-to-atom bonds. Any subsequent dissociation process must involve a change in those bonds — a stretch or a bend, for example. The frequencies of those bond changes are known as vibrational frequencies. The shortest frequencies correspond to a period of about 10–14 s; usually several periods are required as a prelude to dissociation. Ergo, the dissociation occurs much more slowly than the ionization does.
- Ion dissociation reactions occur more slowly than any redistribution of energy among internal degrees of freedom of the ion.
Before any dissociation occurs, an ion has ample time to redistribute energy throughout the rotational and vibrational (and in some models, the excited electronic states) of the ion. These internal redistributions are mostly radiation-less, although there are examples in which fluorescence radiation from organic ions has been observed. Vibrational relaxation can occur within electronic states of the ion, or vibronic (mixed vibrational and electronic relaxation) can occur. In most statistical rate theories, a randomization of energy within all possible modes is assumed, although there have been instances of isolated electronic states. In any case, all of this energy redistribution occurs before formation of the activated complex and before any breaking of bonds that leads to ion dissociation.
- Ions are in a state of internal equilibrium, but do not exchange energy with the environment.
The energy distribution that takes place after ionization but before any dissociation reaction puts the internal energy of the ion into vibrational and rotational states. There is not one single state, but a collection of possible states that are considered in equilibrium. The collection of states might be thought of as the "system" in analogy with the temperature of a classical system. However, the ion does not exchange energy with the outside world. Interactions with the outside world would occur in a collision, but the pressure in the EI source is low and collisions are unlikely before the ion dissociation occurs (these probabilities can be calculated). If a collision were to occur, it could either excite or de-excite the ions. Collisional quenching is a well-known process, and collision-induced dissociation is used in mass spectrometry–mass spectrometry (MS-MS). But within the statistical rate model, and the present discussion, the ion is considered to be an isolated entity. Strictly speaking, the exchange of energy with the external environment could also involve the emission of radiation, which is known to occur in some ions created by EI. The ions that we model are lonely, and they are in a cold, dark place.
- Ionic products are formed by a series of competing, and sometimes consecutive, dissociation reactions.
The rate equation that is developed in these theories includes a symmetry factor that accounts for the possibility that there may be several accessible dissociation reactions with similar activation energies and density of state factors. Figure 1 illustrates one possible reaction path of many that are possible, and the true reaction surface is a more complicated shape. It makes sense that after the internal energy is randomized within the ion, an individual ion can choose to react whichever way it chooses, and a population of ions will exhibit dissociation products from all accessible dissociation reactions. Furthermore, a second dissociation of a product ion can occur, and if the activation energy for the second reaction is low, a mass spectrum may contain only the final end product of a series of dissociation reactions.
Comparing Theory to Experiment
These four separable assumptions can be distilled into one maxim — the ions, once formed, redistribute internal energy (and even structures) in isolation until they reach an equilibrated collection of their most stable state, then (and only then) can dissociation reactions occur. Over the past 60 years as these models have been developed, the test has been to calculate the rates of dissociation reactions from a rate equation and, in doing so, "predict" a mass spectrum. Multiple comparisons of theoretical predictions to experimental results have been performed, and the theories have been modified, refined, extended, and sometimes reformulated. This grand task has been documented in multiple books and reviews (1–4). The importance of the theory and its development are exemplified by the award of a Nobel Prize to Marcus in 1992 for "his contributions to the theory of electron transfer reactions in chemical systems" (5).
The rate of dissociation reactions for an isolated ion with a given internal energy E in a simple form is given by the following equation:
k(E) = (σ/h) [(W‡ (E – E0))/ρ(E)] [1]
where k(E) is the rate as a function of energy E; E0 is the activation energy; s is the symmetry factor representing the number of reactions having the same activation energy and activated complex; h is Planck's constant; W‡ (E – E0) is the number of vibrational and rotational states with an energy less than (E – E0); and ρ(E) is density of states of the reactant ion with energy between E and (E + dE).
The increased rate with an increased number of states and density of states is apparent in the equation. To actually calculate the rate of a dissociation reaction, the number of states and the density of states must be estimated. Two central assumptions underlie the variety of methods used to obtain these values: only vibrational and rotational states are considered, and all of the oscillators used to describe these states are assumed to be harmonic. Isolated exceptions are of course to be found in the studies of ion dissociation reactions, and among the most common exception is the discovery of isolated electronic states. Vibrational and rotational states for the ion are often approximated from the measurable states of the gas-phase neutral molecule from which the ion is formed. Despite the mathematical complexity of some parts of these theories, it makes sense that the metrics considered are those that relate to the stretching and bending of bonds and therefore the structure of the activated complex.
As can be expected, the test between theory and experiment is most valid when the experiment itself is tightly constrained and when the model systems are simple. Accordingly, the results of such comparisons remain an integral part of research in this area. New formalisms are proposed for the theoretical underpinnings, both as new approaches and to address inconsistencies. Lorquet (6) provides perspective on landmarks in the development of theory. Of course, as computational methods and computers improved, these new tools were applied to modeling ion dissociation rates (7,8). A significant shift also occurred as MS developed ionization methods to create ions from previously intractable samples, and as the masses of ions moved higher and higher. Inevitably, researchers want to benchmark the energetics of ions created by these new ionization methods (such as electrospray ionization [ESI]), and to consider whether higher mass ions could be energized to the point of dissociation in the same way as lower mass ions (9). This latter interest arose because the new ionization methods were thought of as "soft," leaving a minimum of internal energy in the ions. Furthermore, researchers thought that internal energy was distributed between a much higher number of states, so that the opportunity to form an activated complex that would subsequently dissociate would be reduced. Accordingly, the link between energy deposition into larger ions by means other than the initial ionization process has been tested through some of the same theoretical approaches.
As examples of recent work in ion energetics and the determination of rate constants, a systematic approach to specifying frequencies in linear peptides has been systematically compiled. Along with consensus conditions for other relevant parameters, RRKM (Rice-Ramsperger-Kassel-Marcus) calculations were shown to be feasible for estimation of reaction rate constants for peptide ions with masses as high as several thousand daltons (10). ESI, in all of its variants, has been a highly charged area of research (11,12). Variations in the mass spectra recorded with different configurations of the ESI source were linked to changes in the internal energy distributions. Furthermore, changes in the composition of the sprayed solution allowed manipulation of the internal energy and rates of dissociations. Once these studies were performed, and the energetic basis established, the use of ESI-MS for observing noncovalent complexes and determination of binding constants was quantitatively developed. As new variants of the ESI technique are developed, their energetic characteristics are also determined (13). Source characterization is a prelude to use of these methods for intricate experiments such as biological imaging and the analysis of single cells, and the deconvolution of the complex mass spectra recorded for such samples (14).
References
(1) P.J. Robinson and K.A. Holbrook, Unimolecular Reactions (Wiley-Interscience, Hoboken, New Jersey, 1972).
(2) J.H. Beynon and J.R. Gilbert, Applications of Transition State Theory to Unimolecular Reactions (Wiley and Sons, Hoboken, New Jersey, 1984).
(3) D.G. Truhlar and B.C. Garrett, Ann. Rev. Phys. Chem.35, 159–189 (1984).
(4) T. Baer and W.L. Hase, Unimolecular Reaction Dynamics, Theory and Experiments (Oxford University Press, New York, New York, 1996).
(5) http://www.nobelprize.org/nobel_prizes/chemistry/laureates/1992/
(6) J.C. Lorquet, Int. J. Mass Spectrom.200(1–3), 43–56 (2000).
(7) W.T. Duncan, R.L. Bell, and T.N. Truong, J. Computational Chem. 19(9), 1039–1052 (1998).
(8) L. Drahos and K. Vekey, J. Mass Spectrom.36(3), 237–263 (2001).
(9) J. Laskin and J.H. Futrell, Mass Spectrom. Rev.24(2), 135–167 (2005).
(10) J.H. Moon, J.Y. Oh, and M.S. Kim, J. Amer. Soc. Mass Spectrom.17, 1749–1757 (2006).
(11) L. Drahos, R.M. Heeren, C. Collette, E. De Pauw, and K. Vekey, J. Mass Spectrom. 34(12), 1373–1379 (1999).
(12) V. Gabelica and E. De Pauw, Mass Spectrom. Rev.24(4), 566–587 (2005).
(13) P. Nemes, H. Huang, and A. Vertes, Phys. Chem. Chem. Phy.14, 2501–2507 (2012).
(14) http://vertes.columbian.gwu.edu/index.php.
Kenneth L. Busch notes that the rates of dissociation reactions of isolated ions are of interest in atmospheric and cosmological chemistry as well as mass spectrometry. The ions found in outer space are usually smaller, and derived from stars, protostars, and the occasional planet, but who can be sure what comes out the tailpipe of a Klingon battle cruiser? This column is the sole responsibility of the author, who can be reached at wyvernassoc@yahoo.com.



Kenneth L. Busch

















LIBS Illuminates the Hidden Health Risks of Indoor Welding and Soldering
April 23rd 2025A new dual-spectroscopy approach reveals real-time pollution threats in indoor workspaces. Chinese researchers have pioneered the use of laser-induced breakdown spectroscopy (LIBS) and aerosol mass spectrometry to uncover and monitor harmful heavy metal and dust emissions from soldering and welding in real-time. These complementary tools offer a fast, accurate means to evaluate air quality threats in industrial and indoor environments—where people spend most of their time.


NIR Spectroscopy Explored as Sustainable Approach to Detecting Bovine Mastitis
April 23rd 2025A new study published in Applied Food Research demonstrates that near-infrared spectroscopy (NIRS) can effectively detect subclinical bovine mastitis in milk, offering a fast, non-invasive method to guide targeted antibiotic treatment and support sustainable dairy practices.

Smarter Sensors, Cleaner Earth Using AI and IoT for Pollution Monitoring
April 22nd 2025A global research team has detailed how smart sensors, artificial intelligence (AI), machine learning, and Internet of Things (IoT) technologies are transforming the detection and management of environmental pollutants. Their comprehensive review highlights how spectroscopy and sensor networks are now key tools in real-time pollution tracking.


