The Use of Collision/Reaction Cell ICP-MS for the Determination of Elements in Blood and Serum Samples
This study describes the development of a robust, high-throughput analytical method for the determination of 18 elements (15 trace elements and three electrolytes) in blood and serum samples using a collision/reaction cell quadrupole ICP-MS system.
The analysis of metals in clinical fluids such as whole blood, serum, and urine has been used for many years to provide information on toxicity, workplace exposure, nutrient availability, and as a diagnostic tool for a number of ailments. The fact that many trace metals are present at variable and often low concentrations (sub-nanogram-per-milliliter range) in different clinical sample types has presented clinical analysts with a variety of challenges. In addition, matrix components, such as organic compounds, proteins, or electrolyte salts that can interfere with the analysis of trace elements often are present at elevated levels (milligrams per milliliter or above). Depending upon the matrix to be analyzed, the amount of sample that can be taken and the means of sampling also can impose restrictions. Sufficient volumes of urine normally can be obtained from patients with non-invasive techniques, whereas the collection of whole blood or serum samples usually involves the use of needle and syringe and generally yields smaller sample volumes (often only microliters or milliliters) for analysis. The analysis technique employed should, therefore, provide the following capabilities — sufficiently low detection limits, the ability to overcome matrix-related interferences, sufficient linearity to measure a wide concentration range in unknown samples, simultaneous multi-elemental determinations, and the ability to cope with small sample volumes.
Analysis of clinical matrices by inductively coupled plasma-mass spectrometry (ICP-MS) is becoming more widespread because ICP-MS meets a number of the above requirements, such as very low detection limits for many trace metals (sub nanogram per milliliter), relative freedom from interferences, simultaneous multielemental determination, and suitability for small sample volumes, as well as providing isotopic information and the possibility of employing isotope dilution mass spectrometry (IDMS) as a high-caliber reference calibration technique. Although some methodologies for the multi-elemental application of the ICP-MS technique to clinical sample matrices have been described in the literature (1–3), the majority of publications have focused on the analysis of single elements in these matrices. Part of the reason for this is that some of the elements of interest suffer from mass spectral and nonspectral interferences derived from the sample matrix when analyzed by ICP-MS (3). Recently, the routine determination of 23 elements in urine samples after fivefold dilution with 1% HNO3 using an octopole-based collision/reaction cell ICP-MS (CRC-ICP-MS) system, was described (4). Historically, matrix-based spectral interferences have been overcome by the use of sector field or high-resolution inductively coupled plasma mass spectrometry (HR-ICP-MS) (5) or by non-MS techniques such as atomic fluorescence (6) or atomic absorption (AA) spectroscopy (7). Another method of overcoming matrix effects has employed the use of sample digestion using concentrated acids or ashing techniques (8). These techniques can be expensive, time-consuming, and less suitable for high sample throughput.
In our laboratory, magnetic sector HR-ICP-MS (Element 1, ThermoFinnigan) has been used (5, 9) for monitoring post- and preoperation samples from patients with metal-on-metal hip replacements. After 1:20 dilutions of blood and serum samples with ~0.7 mM ammonia, 0.01 mM EDTA, and 0.07% (v/v) Triton X-100, or 1:15 dilutions of urine samples with 1% HNO3, elements including Al, V, Co, Cr, Mo, Ni, and Ti were analyzed. The main drawbacks of this technique were cost, lack of practicality, and duration of instrument set-up, as well as instrument downtime and matrix tolerance during analytical runs containing more than ~30 blood or serum samples.
Objectives
The aim of this work was to develop a robust ICP-MS methodology based upon collision/reaction cell quadrupole ICP-MS (CRC-ICP-MS), capable of measuring a wide range of elements in a single analysis after only a simple dilution of the samples.
- A simple dilution of the samples was selected as the preferred sample preparation method because acid digestion techniques can increase the sample turnaround time, cost, and the potential for contamination.
- To achieve the required sample throughput of up to 100 samples per batch, the quantitation method had to be based upon external calibration. Minimal instrument drift, therefore, was paramount to reduce the need for frequent recalibration or drift correction.
- The target elements included the trace metals Al, As, Cd, Co, Cr, Cu, Fe, Mn, Mo, Ni, Pb, Sb, Se, V, and Zn, as well as the electrolytes K, Mg, and Na.
- The sensitivity achieved was required to match previously obtained detection limits using HR-ICP-MS in our laboratory of 0.2 ng/mL (for example, Co and Mo) and 1.0 ng/mL (Ni) in the undiluted samples.
Sample Preparation
All samples, standards, and QC materials were diluted 20-fold using a solution containing ~0.7 mM ammonia (BDH, Poole, UK), 0.01 mM EDTA (Aldrich, Poole, UK) and 0.07% (v/v) Triton X-100 (BDH, Poole, UK). Butan-1-ol (Fluka, Poole, UK) was added to the diluent as a carbon source at 1.5% (v/v) to improve matrix matching between standards and samples and thereby increase the accuracy for analytes such as As and Se whose ionization behaviors in the plasma are affected by the carbon content (10).
To keep the chemistry of the sample introduction system stable throughout the run, the diluent also was used for pre- and postanalysis rinse functions. Use of common-rinse acids such as HNO3, even at dilute levels of 1%, can lead to coagulation or precipitation of clinical matrix components and result in blockages of tubing or nebulizers.
The selection of internal standard elements and the internal standard concentration is very important. The choice of elements often is restricted in clinical analysis due to the presence of many of the elements that usually are used in environmental applications at nanogram-per-milliliter levels in clinical samples. Blood and serum samples were analyzed in semiquantitative mode to determine the most suitable internal standard elements — that is, those that were not present or present at the lowest levels in relative terms. For elements that were present in the samples, such as Sc, the concentration of the internal standard was added at such a level that the contribution from Sc in the sample to the total 45 Sc signal would be negligible.
The internal standard elements (Sc, Ge, Rh, In, and Tl) were chosen according to the best possible match in terms of proximity in mass to charge ratio and ionization behavior and were added to the diluent at a concentration of 20 ng/mL. Addition of the internal standard in this way negated possible mixing problems if on-line addition of the internal standard via a T-piece was used.
ICP-MS Conditions
An Agilent (Palo Alto, CA) 7500ce collision/reaction cell ICP-MS was used in three different gas modes — hydrogen, helium, and standard, or no-gas, mode. The ICP-MS conditions and the isotopes, integration times, and gas modes for the multielemental determination are given in Tables I and II. Quantitation on all isotopes was performed using the three central points of the spectral peaks.
A 100-µL/min PFA microflow nebulizer was used and sample uptake and washout times were reduced using the larger diameter peristaltic pumps of the Integrated Sample Introduction System (ISIS) (Agilent). The pump speed was set at 0.1 rps during the analysis and washout to minimize overloading of the sample introduction system and the plasma with matrix components. The torch was equipped with a 2.5-mm diameter injector and the Shield Torch System was used. Nickel (Ni) cones were used at all times.
The total acquisition time per sample was 208 s. This included the sequential loading of the H2, He, and Std tune files and a 40-s equilibration and stabilization time between the different gas modes. Each sample–standard solution was analyzed sequentially in all gas modes before the autosampler probe moved to the next sample. After each sample, the autosampler probe was rinsed for 5 s using 5% HNO3 and the sample introduction system, then was rinsed using the diluent for 30 s.
Method Performance and Robustness
The stability of the proposed methodology was tested by running blood and serum samples in a sequence over a period of 10 h (a total of 90 samples including calibration standards and QC checks) and monitoring the behavior of internal standard elements, calibration slopes, and check standards.
Instrument stability – signal variation for internal standard elements. Typical signal variation for the internal standard elements of choice (Sc, Ge, Rh, In, and Tl) was 4.8–9.3% in hydrogen mode, 5.5–8.2% in helium mode, and 6.7–10.0% in standard mode. This was assessed during a 90–sample sequence of blood and serum samples. Figure 1 shows the variation for the internal standard elements throughout the 10-h run. Sc is present in some clinical sample types at nanogram-per-milliliter levels, which can be seen here after sample 8.


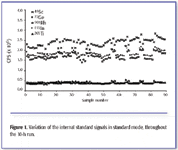
Figure 1. Variation of the internal standard signals in standard mode, throughout the 10-h run.
Calibration repeatability and linearity. The robustness of the calibration technique was assessed by overlaying calibration curves from the beginning, middle, and end of the 10-h run. The correlation coefficients for the mean slope of three calibrations for V, Se, and Pb (Figure 2) during a 10-h sequence ranged from 0.9997 to 1.0000 and indicate the robustness of the method with these matrices. The calibration coefficients for all elements measured were better, generally, than r2 > 0.9900.



Figure 2. Linearity of overlaid calibration curves for V, Se, and Pb, showing stability of the external calibration approach throughout a 90-sample sequence.
Check standards. Check standards at the 1-ng/mL level were analyzed throughout the run after every nine samples for the trace metals and were within 10% of the expected value for the elements tested.
Effects of sample matrix on the sample introduction system. Using dilution factors of 20-fold or less for analysis of these matrices by HR-ICP-MS led to frequent problems with the sample introduction system, especially blocking of the torch injector, which had an internal diameter of 1 mm. When using quadrupole ICP-MS as described earlier, dilution factors of 15- and 10-fold can be used without detrimental effects on the sample introduction system (Figure 3) or instrument performance. The internal diameter of the torch injector used with the CRC-ICP-MS was 2.5 mm. Reagent blanks were monitored after the analytical run, and no significant deterioration in the detection limits or increase in the background levels was observed.



Figure 3. Photos of the interface and sample introduction system after a 90-sample run. Both the sampler and skimmer cones show minor matrix deposits. The 2.5-mm injector torch used was relatively deposit-free. The blood deposits on the spray chamber and the nebulizer block were removed using a sodium hypochlorite solution.
Analysis of Certified Reference Materials (CRMs)
Multiple sub-samples (n = 4) of the certified reference material NIST SRM 1598 Bovine serum and the reference material Seronorm MR9067 (whole human blood, level 2) were diluted 20-fold as described above and analyzed using the conditions described in Tables I and II. These materials were chosen because they represented different clinical matrices and contained a wide range of analytes of interest ranging in concentration levels from sub-nanogram per milliliter to milligrams per milliliter. Levels for the same analytes often varied by more than an order of magnitude between the two materials. Certificate data for both materials, as well as method detection limits (calculated back to the undiluted sample and based upon 3σ of the blank concentration), for the method proposed here are shown in Table III.
The analytical data for both materials were converted to percent recovery data relative to the certified or indicative values and are shown in Figures 4a and 4b. The combination of the reference materials chosen for this study provided certified values with uncertainty estimates for all of the elements determined except for Na, for which only an indicative value was available. The recovery for Na, compared to the indicative value, was 99.0%, and the data for the remaining elements measured fell within the uncertainty range for either one or both of the reference materials. Where the certificate values were not achieved for SRM 1598 (for example, Cr and Cd), the certified concentrations were below the detection limit for the method. Na, As, Ni, V, and Pb are quoted as indicative values only in SRM 1598 (see Table III).



Figure 4. Data obtained expressed relative to the certified data for (a) human blood Seronorm MR9067 and (b) bovine serum SRM NIST 1598. Errors bars represent expanded uncertainties for data obtained and certified ranges for the reference materials. The numbers after the isotopes indicate the tune step used (1 = H2, 2 = He, 3 = Std).
Matrix-matching. The data for As and Se in MR 9067 are slightly high compared to the certified mean value, and this could be due to a higher carbon content in this matrix. When increasing the level of butan-1-ol to in the diluent from 0% to 3% v/v, recoveries for these analytes decreased and approached 100% (Figure 5). When no butan-1-ol was added to the diluent, recoveries for As and Se were significantly higher than the mean certified values (by 94% and 72% respectively) in comparison to recoveries obtained with butan-1-ol addition at 1.5% (v/v). A complete matrix match was achieved for both samples by using the standard addition technique for As and Se in both reference materials. Recoveries for Se were 95.8% and 99.9% in NIST SRM 1598 and Seronorm MR9067, respectively, and 102.6% for As in Seronorm MR9067.


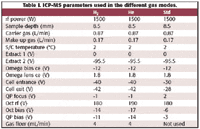
Table I. ICP-MS parameters used in the different gas modes.
Figure 5 also indicates that the effect of the carbon addition on both elements is slightly different. According to the data obtained here, an addition of 3% would be best for As (mean recovery of 95.4 ±2.3%), whereas the ideal volume of butan-1-ol addition for this sample and dilution level for Se is closer to 2%.


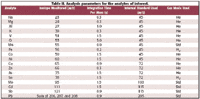
Table II. Analysis parameters for the analytes of interest.
For elements in which the ionization is affected by matrix components in the plasma, it is therefore imperative to obtain a good level of matrix matching for the greatest accuracy. If this is not possible, for example, if the carbon levels in different samples vary significantly, it might be better to use a different sample preparation procedure such as closed-vessel microwave digestion to destroy the organic carbon matrix. However, this can increase the sample turnaround time for large sample batches significantly.


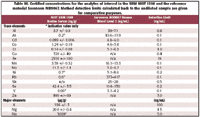
Table III. Certified concentrations for the analytes of interest in the SRM NIST 1598 and the reference material Seronorm MR9067. Method detection limits calculated back to the undiluted sample are given for comparative purposes.
Spike Recovery Data
Spike recovery experiments were performed on both materials for the trace metal analytes at two to four different levels with concentrations ranging from two to five times of the original analyte concentrations. The only exception to this spiking regime was Fe in MR9067, for which no certificate or indicative value was available before the analysis and where the spike concentration added (20 ng/mL) was not sufficiently high above the determined sample concentration (400 ±5 µg/mL) to give meaningful recovery data. The mean data for all spike levels for the trace metal analytes are shown in Table IV. Spike recoveries for all elements fell within 100 ± 20%, and all except Fe, Se, and Mo were within 100 ±10%. The high Se recoveries are thought to be due to the fact that the matrix matching for carbon content consisted of only 1.5% butan-1-ol. High recoveries for Mo also were observed when the samples were analyzed by HR-ICP-MS, and this effect currently is under closer investigation.



Table IV. Mean spike recovery data obtained for both reference materials.
Conclusions
A robust CRC-ICP-MS method has been developed that is capable of high sample throughput (up to 100 samples per batch) for a large suite of elements in difficult clinical matrices after simple dilution. The method robustness has been demonstrated by minimal signal drift during analytical sequences of 10-h duration, negating the need for frequent recalibration. The method detection limits achieved matched those of a previously used HR-ICP-MS method. Further improvements in method detection limits can be achieved by reducing the dilution levels of the clinical matrices, which is possible due to the robustness of the sample introduction system. Good agreement within the uncertainty of certificate values was achieved for all of the target analytes in both reference materials, for which certified data were available across concentration levels ranging from nanograms per milliliter to milligrams per milliter. Spike recoveries for all elements fell within 100 ±20%, and all except Fe, Se, and Mo were within 100 ±10%.


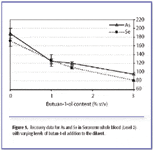
Figure 5. Recovery data for As and Se in Seronorm whole blood (Level 2) with varying levels of butan-1-ol addition to the diluent.
Acknowledgments
The work described in this paper was supported under contract with the Department of Trade and Industry (U.K.) as part of the National Measurement System Valid Analytical Measurement (VAM) program. Agilent Technologies is acknowledged for provision of the CE lens system upgrade on the 7500 ICP-MS for the work described here.
References
1. M.A. Vaughan, A.D. Baines, and D.M. Templeton, Clin. Chem . 37, 210–215 (1991).
2. E. Barany, I.A. Bergdahl, A. Schütz. Skerfving, and A. Oskarsson, JAAS 12, 1005–1009 (1997).
3. C. Sariego Muniz, J.M. Marchante-Gayon, J.I. Garcia Alonso, and A. Sanz-Medel JAAS 14, 193–198 (1998).
4. P. Heitland and H.D. KÖster JAAS 19, 1552–1558 (2004).
5. C.P. Case, L. Ellis, J.C. Turner, and B. Fairman, Clinical Chemistry 47(2), 275–280 (2001).
6. J.A. Holcombe and T.M. Rettberg, Anal. Chem. 58, 124R (1986).
7. A. Taylor and P. Green JAAS 3, 115–118 (1988).
8. L. Dunemann, Nachr. Chem. Tech. Lab . 39(10), M3 (1991).
9. D Ladon, A. Doherty, R. Newson, J. Turner, M. Bhamra, and C.P. Case, J. Arthroplasty 19(8), 78–83 (2004).
10. E.H. Larsen and S. StürupJAAS 9, 1099–1105 (1994).
R. Wahlen, L. Evans, J. Turner, and R. Hearn are with LGC Limited (Teddington, Middlesex, U.K.). E-mail: raimund.wahlen@lgc.co.uk











High-Speed Laser MS for Precise, Prep-Free Environmental Particle Tracking
April 21st 2025Scientists at Oak Ridge National Laboratory have demonstrated that a fast, laser-based mass spectrometry method—LA-ICP-TOF-MS—can accurately detect and identify airborne environmental particles, including toxic metal particles like ruthenium, without the need for complex sample preparation. The work offers a breakthrough in rapid, high-resolution analysis of environmental pollutants.


The Fundamental Role of Advanced Hyphenated Techniques in Lithium-Ion Battery Research
December 4th 2024Spectroscopy spoke with Uwe Karst, a full professor at the University of Münster in the Institute of Inorganic and Analytical Chemistry, to discuss his research on hyphenated analytical techniques in battery research.

Mass Spectrometry for Forensic Analysis: An Interview with Glen Jackson
November 27th 2024As part of “The Future of Forensic Analysis” content series, Spectroscopy sat down with Glen P. Jackson of West Virginia University to talk about the historical development of mass spectrometry in forensic analysis.

Detecting Cancer Biomarkers in Canines: An Interview with Landulfo Silveira Jr.
November 5th 2024Spectroscopy sat down with Landulfo Silveira Jr. of Universidade Anhembi Morumbi-UAM and Center for Innovation, Technology and Education-CITÉ (São Paulo, Brazil) to talk about his team’s latest research using Raman spectroscopy to detect biomarkers of cancer in canine sera.


