USP <1058> Analytical Instrument Qualification and the Laboratory Impact
In this column, Bob McDowall discusses the impact of USP general chapter 1058 on the spectroscopy laboratory.



United States Pharmacopoeia (USP) general chapter 1058 on analytical instrument qualification (AIQ) has finally become official and was released in August 2008 in the first supplement to the USP XXXI (1). In this collumn, I'll revisit this approach to AIQ and discuss the impact that this will have in the regulated spectroscopy laboratory.
In my 2006 column (2), I wrote about the draft USP General Chapter 1058, or USP <1058> on analytical instrument qualification (AIQ). Now that USP <1058> is effective from August 2008, I want to revisit the subject and look in more detail at the approach and the impact this will have on the regulated spectroscopy laboratory.



R.D. McDowall
As a reminder, USP <1058> originated at a conference organized by the AAPS (American Association of Pharmaceutical Scientists) in 2003 entitled "Analytical Instrument Validation." The first thing to bite the dust was "validation" because the attendees agreed that instruments are qualified and processes, methods, and computer systems are validated. Thus, analytical instrument qualification was born. Prior to this it had humble beginnings as simply equipment qualification (EQ). The debate about EQ versus AIQ should be reserved for a bar with copious amounts of alcohol because you will be talking about the same process except the name has changed. Either term means that any instrument is fit for its intended use — nothing more and nothing less.
AAPS published a white paper of the conference in 2004 (3), incorporation as a potential USP general chapter followed in 2005, and review cycles followed until it was finally adopted. It is worth remembering at this point that USP general chapters between <1> and <999> are requirements (you must comply or your product may be deemed adulterated) and those between <1000> and <1999> are informational in nature (alternative approaches are OK if they are justified).
Take care, however. Although <1058> is informational, it implicitly refers to other USP GENERAL CHAPTERS pertinent to specific techniques (for example, <21> Thermometers or <41> Weights and balances that are requirements). Therefore, be warned and careful in your overall approach to qualification.
Potential Problems with USP <1058>
At first sight, USP <1058> seems fine: it appears to be a logical and practical approach to qualification of equipment and instrumentation. It is only when you dig into the details and explore the implications that you find that there are potential problems that could catch you out if you don't think things through and interpret the general chapter sensibly. As we go through this column, I will point out areas that will need special care in your interpretation and approach.
Positioning AIQ versus Other Laboratory Quality Checks
A common debate with analysts who do not understand the problem is that if we validate an analytical method it does not matter which instrument we run it on: a spectrometer is a spectrometer, isn't it? Unfortunately, this is not the case, and a hierarchy of quality checks is described succinctly in <1058>.
The foundation of all quality analytical work is the qualification of the instrument: so you do undertake AIQ first (ideally during the purchase and installation and before you use the instrument). This establishes that the instrument is fit for use around the operating parameters that you test against.
Similar to building a house, the next stage is to develop and validate analytical methods for your work within the parameters you have qualified — don't exceed them, as your instrument is not qualified.
Next, when you apply the method, you'll check that the instrument works before you commit samples for analysis. This could be a balance check versus a known calibrated weight to see correct operation or that a spectral library can identify known standards. This is the equivalent of a system suitability test, or SST, which is typically performed on the day you analyze the samples.
Finally, you will include quality control and perhaps blank samples to check that the instrument readings are OK and give you confidence in the method's operation on the day.
This is shown diagrammatically in the USP chapter as a triangle with AIQ at the base and QC samples at the apex.
So the bottom line is if you don't qualify the instrument or do it incorrectly, all the other work you do is potentially wasted. So that's the easy bit, but the problem with <1058> is that it is written from the perspective of the laboratory. But do you develop and manufacture your own instruments? Probably not, so let's look in more detail at the basics of the instrument qualification process described in <1058>. In doing so, we'll revisit the hierarchy of the quality checks again but in an expanded form.
Instrument Qualification Process
The process for instrument qualification follows the 4Qs model for the specification, installation check, and monitoring of ongoing instrument performance.
Design Qualification (DQ): Define the functional and operational specifications of the instrument and any associated software for the intended purpose. To be performed before purchase of a new instrument.
Installation Qualification (IQ): Establish that an instrument is delivered as designed and specified and that it is properly installed in the selected environment. To be performed at installation of the system on new and old or existing systems (please see my comments below).
Operational Qualification (OQ): Document that the instrument will function according to its operational specification in the selected environment (so what happened to defined intended purpose?).
Performance Qualification (PQ): Document that the instrument consistently performs according to specification and intended use. I will not discuss PQ further in this column.
However, there are a few little problems with this simplistic approach.
I'm lazy — let's get the vendor to do the DQ.
This is one of the major causes of trouble with this approach. Under DQ the chapter states that, "DQ may be performed not only by the instrument developer or manufacturer but also may be performed by the user." Wrong! Wrong!! Wrong!!! A manufacturer's specifications have a number of purposes, such as to show that their instrument is better than the competition, to limit the vendor's liability, and to be the benchmark to compare against when the instrument is installed. Furthermore, in some cases, a manufacturer's specifications may be totally irrelevant and meaningless to your intended use. As a specific example, a benchtop centrifuge had a rotor speed specification of 3500 ± 1; however, on further investigation this was measured without a rotor. Therefore, what manufacturer's instrument specifications are not useful for is your intended use specification or to be the sole component of an instrument's DQ. This is the total abrogation of the user's responsibility.
You, the laboratory user, are responsible by law under the GMPs to define intended purpose for your instruments. Furthermore, you, the laboratory user, are responsible to your company to purchase instruments that meet your business need. Never rely on a manufacturer's specification to specify your requirements; however, it can be used to enhance or change your specification.
More Detail of the Quality Checks Hierarchy
The <1058> triangle that I mentioned earlier must be modified so that you can have a better understanding of my approach to the 4Qs model. My take on this is shown in Figure 1, and it is shown as a time sequence running from left to right. The elements of data quality described in <1058> are shown as the top four layers. I have expanded the AIQ layer to show how it relates to the instrument vendor's processes, and we will concentrate on these two layers in more detail.


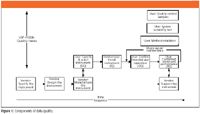
Figure 1
In the beginning was an instrument that was specified, designed, manufactured, and supported by a vendor to perform one or a series of analytical tasks. This instrument sits at the vendor's facility until a salesperson visits, or you flick through an issue of Spectroscopy, drop into an exhibition, or decide to purchase an instrument to do a specific task.
STOP. Do not pass go and do not collect $200. Also, do not get seduced by the salesperson slithering over the floor toward you. Here is where you have to do some work and define what you want the instrument to do, especially if it is a new technique for your laboratory. Do some leg work and research and define the following:
Key operating parameters?
Sample presentation to the instrument: liquid, solid, semi-solid?
Numbers of samples you expect: do you need an autosampler?
Bench space available?
Services: Power supply required and any other services?
Environment: Will you be analyzing toxic substances, or can the system sit in the laboratory?
Now write it down: this is your specification. Then approve it, refine it, and update it because it's a living document.
GO. Now check your specification versus the instrument from a vendor because you have the basis on which you can make the decision: your specification. This is why <1058> is wrong to suggest that the bulk of the work is a vendor's; it is not. Only a user can undertake a full DQ. A vendor might supply their specification, but that is only to compare with yours. Ignoring your specification is a high business risk, but you have never bought an instrument that did not work, have you?
Look outside the pharmaceutical industry for a moment to other quality standards. ISO 17025 has a footnote to laboratory equipment that most people gloss over, but it is very important in this context. The footnote states that laboratory and manufacturer's specifications might be different; you might want the system to do something that a manufacturer has not considered or work in a very narrow range of the spectrum that the instrument is capable of. Hence, the need to define your instrument and system requirements, otherwise you will have problems in that the purchased system might not meet your needs. The message for the DQ phase of AIQ is for the user to ask what they want the instrument or system to do and document it, regardless of the statements in USP <1058> that are designed to dump the responsibility on the vendor and not the user.
Execute a Retrospective IQ?
The section of USP <1058> on installation qualification mentions that IQ also applies to "any instrument that exists on site but has not been previously qualified." I disagree. What this means is that you take your functioning system apart and rebuild it? Now that's an added-value activity that will impress your boss at appraisal time. Consider the following approach as an alternative: what you need to do is establish control over the existing instrument and all its constituent components and show that it works as intended.
First, list all the major components of the system, take the model and serial numbers of the instruments, and details of any computer and the software (application, database, and operating systems and the respective service packs). Of course you might already have this information in a log book, in which case check that the items are correct and update anything that needs it.
Second, collate all documentation associated with the system: installation and service records, and establish, if you have not done so, an instrument/system log book.
Third, put the system under change control and do not change or update any component unless you have gone through a change request.
Fourth, document the fact that no retrospective IQ will be performed because you will be taking a working system to pieces and rebuilding it.
Fifth, carry out a full operational qualification to demonstrate that it performs its intended purpose against your specification.
Now having discussed the problems of the <1058> 4Qs model, let us look at the classification of instruments and equipment.
<1058> Instrument Classification
The problem is that when we look around the laboratory, we see equipment and instruments that vary from a vortex mixer to a complex and sophisticated spectrometer. One question that is raised often is should we apply the same approach to all equipment and instruments in our laboratories? The answer is "no," and the USP <1058> answer is relatively simple and straightforward because all equipment, instruments, or systems are classified into one of three categories:
Group A
Group B
Group C
Table I shows the criteria for classification and how each are qualified, together with some typical examples in each group. There is a built-in risk assessment because Group A is the lowest risk requiring the smallest amount of work and Group C represents the greatest risk and hence, the most work to control.


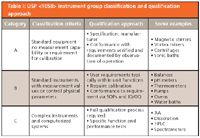
Table I: USP instrument group classification and qualification approach
It's Only a Sonic Bath?
Now let's take an example from the list of instruments and equipment in the right-hand column, say a humble sonic bath, and walk through the classification process and look at the impact. According to <1058>, the bath falls into category A because it is standard equipment and conformance with requirements can be verified by observation of the operation. This approach is OK, isn't it? Well let's look at the small print here. In Table I it says: "Conformance with requirements verified and documented by observation of operation." Note the word requirements. This is your specification, and therefore the classification of the sonic bath as a Group A instrument depends entirely upon what you want to use the instrument for. So if all you are using the sonic bath for is simply dissolving material in volumetric flasks and you will use observation as the way to determine if the compound has dissolved, this is OK. If there is material still undissolved, then all you do is replace the flask in the bath and turn on the power for another minute or so. Yes, the sonic bath used in this way falls into Group A and can be qualified as outlined in Table I.
However, if the method calls for a specific amount of sonic energy and the flasks must be placed in specific locations in the bath, then this is a different intended use that now takes the not-so-humble sonic bath into Group B. Here we need to consider calibration of the bath and map the sonic energy levels and also IQ/OQ to see that the installed unit meets our specification, which of course we will have documented before we bought the bath.
OK, let's take a slightly different tack now and consider our sonic bath again. What would be the situation when it is used as a standalone item of equipment but integrated and used within a robotic system. What do we do now? We need to consider qualification from two perspectives: the bath as a standalone and then integrated within the robotic system. Again, what the bath will be used for in the robotic system will determine the approach, but there will be two qualification passes: a first pass to install and check out the bath itself and a second pass in which the bath will be qualified as part of the operation of the whole robotic system. So the sonic bath would be Group A or B (as discussed earlier) for the first pass qualification and Group C as the second pass robot qualification.
The Importance of Your Requirements
So please do not look at the list of equipment and systems in Table I and in <1058> itself and think that because a particular item falls into a specific category it is a definitive statement. Think about how you will use this and look at your requirements. The one point that all three sonic bath scenarios make is that if you have not defined what it will do, you will be noncompliant with 21 CFR 211.63 (ref. 4):
Equipment used in the manufacture, processing, packing, or holding of a drug product shall be of appropriate design, adequate size, and suitably located to facilitate operations for its intended use and for its cleaning and maintenance.
Writing the specification for laboratory systems, instruments, and systems is the area in which most organizations fail because the entire qualification approach is the 3Qs model of IQ, OQ, and PQ. There is zero consideration of DQ until the users have realized they have bought the wrong instrument!
Don't Validate Software According to USP <1058>
Virtually all instruments have software varying from firmware for the basic instrument operation of some Group B instruments to a separate data system running on a PC to cover instrument control, data acquisition, analysis, and reporting for the Group C instruments. The <1058> approach for Group B instruments is to test the firmware implicitly as part of the IQ and OQ phases of the qualification — this is a good and pragmatic approach that I fully agree with. However, it contrasts with the latest version of the GAMP Guide (5), but we will discuss this subject in more detail in the next "Focus on Quality" installment.
Group C instruments are where I have a difference of opinion with USP <1058> and its approach to software validation. To say that the CSV approach advocated by <1058> is too simplistic and naïve is the politest thing that I can say before the editor reaches for the delete key. For instrument control, acquisition, and processing software, the rationale is to leave the whole job to the vendor and implicitly test during instrument qualification. To quote <1058>: "The manufacturer should perform DQ, validate this software and provide users with a summary of the validation." Dream on, dear reader! "At the user site, holistic qualification, which involves the entire instrument and software system, is more efficient than modular validation of the software alone. Thus, the user qualifies the instrument control, data acquisition and processing software by qualifying the instrument according to the AIQ process."
Let's start looking at this approach for Group C instruments in more detail.
The vendor will validate the software, contrasts with EU and US GMP or GLP regulation in the world that states it is the user's responsibility to validate the system. The key to the problem is, does your use of the system match that of the vendor?
The vendor's software development materials are proprietary and can be seen only when you conduct a vendor audit at the company facilities.
Compare the marketing literature with the software warranty. The marketing literature says the software will do the job, but the warranty states that if anything goes wrong, the vendor is not responsible because the software is used "as is" and the user takes full responsibility for malfunctions. Try telling an inspector this one!
The assumption is that the software will be used as-is and without modification. For nonconfigured or noncustomized applications, this might be true, but for many applications, this is not the case because the software will be modified or adapted by the user. For example, the <1058> approach is totally inadequate and deficient for complex systems because it omits the following critical items of software validation:
- Configuration of security and access control for the user community
- Configuration or customization of the software to match your ways of working
- Definition and testing 21 CFR 11 functions as used in your laboratory
- Implementation of custom reports
- Implementation of custom calculations
- Macros defined and written by the users
For Group 3 instruments with data systems, you will leave yourself exposed if you follow the computer validation approach in USP <1058>.
There is also reference to the FDA guidance for General Principles of Software Validation (6) for the validation of LIMS. While this guidance is probably the best that the FDA has written on software validation, you have to remember that the originating Center is CDRH (Center for Devices and Radiological Health). It is also written primarily from the perspective of developing software for a registered medical device and as such, avoids the use of IQ, OQ, and PQ, which will make the mapping of <1058> to the approach outlined in the guidance much more difficult.
Summary
I have reviewed key parts of USP <1058> on analytical instrument qualification to highlight areas in the general chapter where care will need to be taken to prevent an interpretation that is too literal and which can result in regulatory noncompliance.
R.D. McDowall is principal of McDowall Consulting and director of R.D. McDowall Limited, and "Questions of Quality" column editor for LCGC Europe, Spectroscopy's sister magazine. Address correspondence to him at 73 Murray Avenue, Bromley, Kent, BR1 3DJ, UK.
References
(1) United States Pharmacopoeia, General Chapter <1058> Analytical Instrument Qualification, First Supplement to USP XXX1 p3587 (2008).
(2) R.D. McDowall, Spectroscopy 21(4), 14–30 (2006).
(3) AAPS White Paper on Analytical Instrument Qualification, AAPS 2004.
(4) FDA 21 CFR 211: Current Good Manufacturing Practice Regulations
(5) Good Automated Manufacturing Practice guidelines, version 5 (2008), International Society for Pharmaceutical Manufacturing (ISPE).
(6) General Principles of Software Validation, FDA Guidance for Industry, 2002.








Measuring Soil Potassium with Near-Infrared Spectroscopy
January 8th 2025Researchers have developed a novel three-step hybrid variable selection strategy, SiPLS-RF(VIM)-IMIV, to enhance the accuracy and efficiency of soil potassium measurement using near-infrared spectroscopy, offering significant advancements for precision agriculture and real-time soil monitoring.






