Chemically Selective Imaging with Broadband CARS Microscopy
Spectroscopy
September 2006. The authors rapidly acquire complete vibrational spectra in the fingerprint region using a single femtosecond laser for broadband coherent anti-Stokes Raman scattering (CARS) microscopy to image spatially variant compositions of condensed-phase samples.



Recently, there has been great interest in a variety of novel ultrafast coherent anti-Stokes Raman scattering (CARS) methods that utilize broadband lasers (1–3), often involving narrowband probe pulses (4–6), chirped pulses (7,8), or fiber-broadened pulses (9–11) to obtain broadband vibrational information from molecules. With the variation of single-pulse, broadband, multiplex CARS microscopy discussed here, broad bandwidth spectra are acquired rapidly during the microscope scan and the spatially variant compositions of samples composed of multiple components are readily determined.
The essential feature of this method is the use of a single, ultra-broadband femtosecond (fs) pulsed-laser oscillator, which simultaneously provides all of the photons for the CARS scattering. By a combination of phase and polarization control of the 10-fs laser pulses, complete broadband spectral images are acquired in the vibrational fingerprint regime. An important advantage of this method is that full chemical composition analysis is obtained in a single microscope scan, rather than the multiple scans that are necessary with narrowband CARS microscopy (12–17). There are, however, tradeoffs necessary to gain this spectral width. With narrowband CARS, the signal sensitivity and signal acquisition rate can be much greater, but with broadband CARS, the spatial locations of multiple species can be obtained in a single scan. In the discussion that follows, the techniques of broadband CARS are discussed and illustrated with examples of multicomponent chemical imaging.
In general, various fluorescence microscopy techniques have become popular for highly sensitive imaging, but most methods require the introduction of fluorescent probes that can perturb the system of interest, and are often limited by photobleaching. On the other hand, infrared (IR) and Raman spectroscopy are excellent tools for identifying the chemical properties and physical environments of molecules within a condensed phase sample based upon intrinsic molecular vibrations. However, there are significant limitations to utilizing these techniques for microscopy. The long wavelengths required for IR absorption result in low spatial resolution, and spontaneous Raman scattering has a characteristically small cross-section. Also, sample fluorescence often overlaps the Stokes lines typically used in spontaneous Raman scattering spectroscopy. CARS has the potential to become a powerful spectroscopic tool due to the stronger signal levels of vibrational spectral information and three-dimensional sectioning capability (18).
The vibrational fingerprint region extends from about 800 cm–1 to 1800 cm–1 (19). Therefore, acquiring a full vibrational spectrum with ~1000 cm–1 of bandwidth in this spectral region at each spatial location in a sample would enable discrimination of many sample components simultaneously, even those with similar spectral features (9). Furthermore, it is desirable to obtain this full spectrum in single-pulse mode, without scanning over each frequency region individually.
With the goal of imaging complex samples in mind, several groups have developed highly refined methods to increase the sensitivity of broadband CARS spectroscopy to combine it with microscopy as a label-free, chemically selective imaging technique (1–4,6–11). However, due to the time it typically takes to collect the full spectrum, to our knowledge, only a few groups have constructed microscope images with a CARS spectrum at each pixel (6,9,11).
Many applications would benefit from a sensitive imaging technique that provides chemical selectivity for multiple components simultaneously. For example, it would be valuable to obtain the spatial distribution of specific molecules throughout living biological cells and tissues. In particular, the segregation of lipids into distinct domains within the outermost cell membrane is an area of active investigation with implications for an understanding of protein sorting, cell–cell interactions, and signaling processes (20). Recently, narrowband and multiplex CARS microscopy has been used to image the distribution of two different lipids in model lipid membranes by tuning to a single vibrational band or spectral region of interest (6,14). By increasing the spectral bandwidth, it may be possible to determine the precise chemical composition of such domains without extrinsic chemical markers, which are usually necessary to discriminate between different species in a sample. Recent work in narrowband CARS imaging of living cells (11,16,17) makes the prospect of sensitive broadband CARS for imaging biological systems also potentially exciting.
Another area of great interest for chemically selective imaging is that of chemically amplified polymer photoresists, used in the manufacturing of patterned semiconductor chips for electronics and computing. These photoresists are composed of two similar polymers in a pattern with few hundred nanometer spatial domains of each component. It would be advantageous to have a technique that could analyze these patterned photoresists quickly and easily for quality control purposes. Narrowband CARS microscopy has been used successfully to image single spectral peaks across model polymer photoresists (15), but to quantify multiple materials within the resist at the same time, a broader vibrational spectrum is desirable at each point in the microscope image.
CARS
CARS is a three-photon scattering process in which two intense laser fields, called the pump and Stokes fields, interact with sample molecules to induce coherent vibrations, and subsequent interaction with a probe pulse then results in the emission of a coherent fourth field at a higher frequency. This signal is strongly enhanced when the energy difference between the pump and Stokes pulses matches the energy of an intrinsic molecular vibrational level. Typically, the pump and probe pulses and the Stokes pulse are provided by two separate, synchronized picosecond lasers or two beams produced by a single laser. Single vibrational resonances can be probed one at a time with narrowband CARS microscopy if the difference between the pump and Stokes beams from the picosecond (narrowband) lasers is tuned to match these vibrational frequencies (18).
By incorporating a broadband laser into this scheme, such that the broadband laser provides multiple Stokes frequencies simultaneously and a narrowband laser provides the pump and probe beams, a broad spectrum can be acquired simultaneously (multiplex CARS) without tuning over the individual resonances (Figure 1) (4,6,18). However, due to the high peak-power laser field applied to the sample, in addition to this vibrationally resonant CARS signal, purely electronic third-order frequency mixing processes in the sample also lead to a large nonresonant background across the entire spectrum (2,18,21). Because this nonresonant background results from the interaction of the laser field with the sample of interest, creative manipulations are necessary for suppression or elimination of this nonresonant background so that the much smaller intensity resonant spectrum can be obtained (1,3,4,13).


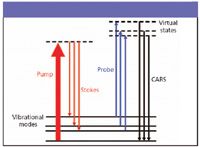
Figure 1: Energy level diagram of the multiplex CARS process using broadband pump and Stokes fields with a narrowband probe field.
With phase and polarization pulse-shaping techniques, multiplex CARS spectroscopy can be accomplished with a single laser beam from an ultrafast laser, eliminating complications associated with tuning, timing, and alignment between two separate beams, and significantly reducing or eliminating the nonresonant background (1). The whole array of pump and Stokes frequencies is provided simultaneously by the broadband pulse, and the resulting coherent molecular vibrations are probed by a narrow spectral part of the total bandwidth within the same laser pulse. The probe part of the beam is differentiated by shifting the phase and polarization of that narrow spectral region of the laser pulse. Fingerprint-region CARS spectra with up to 700 cm–1 of bandwidth have been obtained previously using single-pulse methods (1,3). With a variation of a pulse-shaping and signal-acquisition scheme developed recently in our laboratory (21), sensitivity improvements have been achieved that allow spectral images to be obtained in a shorter acquisition time with up to 1000 cm–1 of bandwidth per spectrum (22).
Spectral Interferometry CARS
The new spectral interferometry method allows the real-time subtraction of the nonresonant background, after it has been utilized to amplify the resonant CARS signal, thus increasing the sensitivity of the single-pulse CARS spectroscopy (21). This allows rapid acquisition of spectra so that images of the chemical distribution in a sample can be constructed based on the spectrum at each point in the sample (22). In this adaptation, the phase and polarization of the narrow probe part of the laser bandwidth are shifted with respect to the rest of the laser spectrum (Figure 2), and two spectra are recorded simultaneously in two different polarization directions. By taking the difference of the two, the desired background-subtracted Raman-equivalent spectrum is obtained directly.


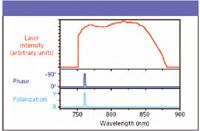
Figure 2: The intensity profile of the laser used in this experiment is shown in red, and the phase and polarization profile is shown in blue. Note that both the polarization and phase of a narrow spectral region (~2 nm) are offset by 90° with respect to the rest of the spectrum to serve as a narrowband probe.
Figure 3 shows the experimental system used for the broadband CARS instrument. A Ti:sapphire oscillator with 10-fs pulses, operating at a 90 MHz repetition rate, provides pulses with ~120 nm of bandwidth. The beam is expanded and then passes through an all-reflective 4f geometry pulse shaper. To shape the phase and polarization of the pulses, the light is first dispersed with a 600-grooves/mm diffraction grating and then passed through a spatial light modulator using a curved mirror. The spatial light modulator consists of two banks of 128 liquid crystal pixels whose birefringence is controlled by changing the applied voltage to the individual pixels.


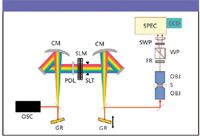
Figure 3: Experimental system: OSC = oscillator, CM = curved mirror, GR = grating, POL = polarizer, SLM = spatial light modulator, SLT = slit, OBJ = microscope objective, S = sample, FR = Fresnel rhomb (achromatic half-wave plate), WP = Wollaston prism, SWP = sharp-edge short-wave pass filter, SPEC = imaging spectrometer, CCD = two-dimensional charge-coupled device.
After phase and polarization shaping, the pulse is again collimated and the light is focused through a 1.2-NA water-immersion microscope objective into a condensed-phase sample on an xy piezoelectric translation stage parallel to the experimental table. After interaction with the sample, the light is collected through a 1.0-NA water-immersion objective and the laser frequencies are filtered out with a sharp-edge short-wave pass filter. The polarization of the light is rotated by 45° by an achromatic half-wave plate, and the signal is split spatially into two separate directions using a Wollaston prism. Both signal traces are focused into a spectrometer and the resulting spectra are imaged onto a two-dimensional CCD detector.
The total CARS signal is given by


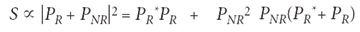
where PR and PNR are the resonant and nonresonant transient polarizations, respectively, which give the CARS signal. The nonresonant contribution is entirely real, but the resonant part of the signal contains both real and imaginary parts. Also, the purely resonant part of the total signal PR* PR is the smallest term, while the nonresonant part PNR2 is the largest term. The cross-term is linear in the nonresonant contribution, but also contains the real and imaginary parts of the resonant contribution. The technique used here detects this larger cross-term, in which the nonresonant signal effectively amplifies the resonant signal (21).
The nonresonant background and resonant CARS signals are coherent, and while the resonant signal depends upon the phase of the probe part of the pulse, most of the nonresonant signal is produced by the broadband part of the pulse, which has a different phase. Thus, it is possible to implement interferometry between the resonant and nonresonant parts of the signal by controlling the phase difference between them. Inducing different phase shifts in the probe part of the pulse with respect to the broadband part results in different interference patterns between the resonant and nonresonant signals that are acquired simultaneously (21).
To directly obtain the resonant CARS spectrum using broadband pulses, the nonresonant background must be eliminated. By rotating the polarization of the probe part of the pulse, the signal can be separated into two traces based upon the differences in polarization between different signal components (Figure 4a). After passing through the sample, the signals at both of these polarizations contain the same nonresonant background, but by inducing a phase offset of –90° in the probe, an equal but opposite interference results between the resonant and nonresonant signal in the two spectra (i and j) at the vibrational resonances, which is shown in Figure 4b for toluene. Note that these two spectra are acquired simultaneously. By subtracting one spectrum from the other, most of the nonresonant signal is subtracted in real time and only the imaginary part of the resonant CARS spectrum remains (Figure 4c). This method is called double quadrature spectral interferometry (DQSI) CARS (21). Note that the nonresonant signal from the sample itself serves as the local oscillator here, which amplifies the resonant signal significantly, providing a sensitivity improvement over previous CARS spectroscopy techniques in which the nonresonant signal is suppressed before detection (2,3,13). DQSI was first developed by Lepetit and colleagues (23) and has been used for CARS previously, but usually by acquiring two spectra under separate conditions (5,12,24). The major benefit here is that there is no need for an interferometer, because the interferometry takes place within the single beam.


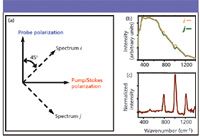
Figure 4: (a) Polarization directions of pump/Stokes (x) and probe (y) fields and two-spectrum detection scheme. (b) Spectra i and j for a probe phase of â90°. The relative intensity of the resonant signal to the nonresonant background is evident in this figure. (c) Difference between spectrum i and spectrum j, which gives the Raman-equivalent (imaginary) part of the resonant CARS spectrum.
Spectroscopy and Imaging
Figure 5 shows DQSI CARS spectra of several common solvents, including toluene, acetone, methanol, ethanol, and isopropanol. The acquisition time for each spectrum is 10 ms. For each sample, the excitation volume is estimated to be about 40 aL (attoliters). The spectral resolution is about 35 cm–1 , which is slightly worse than the limit of 30 cm–1 imposed by the use of two pixels of the dual 128 pixel SLM. All spectra acquired spanned from 400 cm–1 to 1500 cm–1, although only the spectral window of interest is displayed in Figures 5–7.


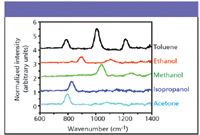
Figure 5: Spectra of some common laboratory solvents obtained with double-quadrature spectral interferometry CARS.
A solution of n,n-dimethylformamide (DMF) was placed on top of the patterned polymer polydimethylsiloxane (PDMS), and the sample was sandwiched between two glass coverslips for imaging. Thin-films of polymethylmethacrylate (PMMA) and polystyrene (PS), which are known to phase separate (25), also were prepared by dissolving 1:1 PMMA–PS in chloroform and spin-casting the solution into a film approximately 2–5 μm thick (thickness verified with atomic force microscopy [AFM], data not shown).
The samples are placed perpendicular to the laser beam (parallel to the table) and scanned through the laser focus in the x and y directions with the piezoelectric translation stage. A spectrum is obtained in 10 ms for each pixel in the images. Each image is 100 X 100 pixels, containing a total of 10,000 separate spectra. For the PDMS–DMF sample, the total image size is 30 X 30 μm, and the PS–PMMA image is 20 X 20 μm. The total time for data acquisition and processing is about 3.5 min for each of the images shown. With current state-of-the-art spontaneous Raman microscopy systems, it typically takes about 1 s to acquire each spectrum, which would result in a total imaging time of 3 h for a scan of similar size and resolution (26).
Spectra of PDMS and DMF are shown in Figures 6a and 6b. The PDMS exhibits a strong band at 460 cm–1 while the DMF exhibits a peak at 830 cm–1. Because each of these peaks is only present in the spectrum of one molecular species, the 460-cm–1 peak specifically identifies the location of PDMS, and the 830-cm–1 peak only appears where there is DMF. Figure 6c shows the analysis of the distribution of these two spectral peaks across the sample. It is clear from the spectral data that the two components are separated spatially into distinct domains and do not overlap in the sample.


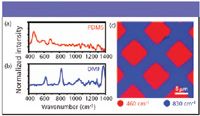
Figure 6: (a) CARS spectrum of pure PDMS. (b) CARS spectrum of pure DMF. (c) Image constructed by taking the spectrum at each point across a patterned sample of PDMS and DMF. Only the 460 cmâ1 (red, corresponds to PDMS) and 830 cmâ1 (blue, corresponds to DMF) peak intensities are plotted in this image.
Figure 7 shows the results for the PS–PMMA spin-cast film sample. Again, the spectra for the individual components are shown (Figures 7a and 7b). However, because the spectra for the two polymers have similar features around both 1000 cm–1 and 800 cm–1, we analyzed the ratio of the these peak intensities for each pixel in the image. The contrast scale for the image in Figure 7c is shown. It is a false-color image in which the bright red corresponds to 1000 cm–1 /800 cm–1 = 4, which is the ratio of the spectral peaks in the pure PS spectrum, and the dark blue is 1000 cm–1 /800 cm–1 = 0.5, corresponding to pure PMMA.


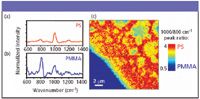
Figure 7: (a) CARS spectrum of pure PS. (b) CARS spectrum of pure PMMA. (c) Image constructed by taking the spectrum at each point across a spin-cast mixture of PS and PMMA. The image contrast is based on the 1000/800 cmâ1 peak ratio. Note that a ratio of 0.5 corresponds to the PMMA spectrum (blue) and a ratio of 4 signifies that mostly PS is present (red). Here, one can also see the relative amounts of both components in the regions where they are mixed.
The spatial resolution in Figure 6 and Figure 7 is about 400 nm, which is less than ideal. For 800-nm light, the best spatial resolution observed to date is 230 nm due to the high numerical aperture of the focusing objective (1.2 NA) and the third-order nonlinearity of the CARS signal, which gives an enhancement of the spatial resolution over a one-photon process (27). The degradation of the spatial resolution is most likely the result of chromatic and achromatic aberrations and misalignment through the focusing objective, which can be significant due to the exceptionally broad bandwidth of the spectrum. Due to the increased demand for near-IR applications in microscopy, newer microscope objectives have improved aberration compensation capabilities in the region of interest to us (28). This should allow for significant improvements in the future.
In addition to imaging multiple species simultaneously, there are two main advantages to taking a spectrum at each pixel and doing a peak ratio analysis for chemical contrast. First, as seen with the PS and PMMA sample, in mixed samples there are often overlapping peaks in the spectra of the molecules of interest. By looking at peaks individually, such as in the first example of PDMS and DMF, true chemical selectivity cannot be achieved for samples whose components have the same or similar spectral features. Second, components in a sample are not always separated into spatially distinct domains, but often are mixed. Using a peak ratio contrast mechanism gives information about the relative proportions of all components simultaneously at each pixel in the image. For example, in Figure 7c, the red regions are pure PS, and the dark blue regions are pure PMMA, but the light yellow and light blue regions in this figure correspond to mixtures of the two polymers. It is evident that the polymers separate into domains tens to hundreds of micrometers in size, but with smaller islands of pure and mixed composition interspersed within these larger domains. AFM images of these spin-cast PS–PMMA films (not shown) indicate that separate regions also differ in thickness from 50 nm to 700 nm, depending upon the initial concentrations of the polymers in solution. For more complex samples, one can utilize the entire spectrum rather than fitting just two distinct peaks, as was done here.
This type of separation is especially prevalent in the outer lipid bilayer membrane of biological cells, where it is hypothesized that different types of lipids segregate into chemically and physically distinct domains. Questions remain as to the size and nature of these domains, but no direct chemical verification of their composition has yet been achieved in living cells, although there is considerable indirect evidence of their properties (20,29). Single-frequency (narrowband) and relatively narrow bandwidth multiplex CARS methods have been used to image the distribution of two specific lipids in model lipid membrane systems simultaneously (6,14).
Sample damage due to three-photon absorption and subsequent local heating is a limiting factor in several of the samples that we imaged. Samples in solution or surrounded by water show increased resilience to photodegradation, but further sensitivity improvements still are necessary to obtain spectral images of polymer photoresists and vesicles of lipid bilayers without causing sample damage. The images shown above were taken with polymer samples that were several micrometers thick, while some polymer photoresists are less than 1 μm thick, and the cell membrane is only 5–10 nm thick. We estimate that two orders of magnitude greater sensitivity is required before this technique will be capable of acquiring a distinct spectrum in just 10 ms from a model membrane composed of a single lipid bilayer, and currently we are incorporating improvements to achieve this. In addition, we are implementing a higher resolution SLM and a laser with increased bandwidth that will extend the accessible spectral range to about 4000 cm–1. This will allow us to see characteristic Raman bands, such as the carbonyl stretch (~1700 cm–1) and CH vibrations (~2800–3000 cm–1) (19), which are prevalent in biological samples, at a higher spectral resolution than previously possible.
CARS microscopy already has paved the way for new types of studies, whereby direct chemical information is obtained on interesting and relevant time and length scales without the need for labeling or molecular markers. Although this technique alone will not be able to achieve single-molecule sensitivity, the direct chemical selectivity of broadband CARS for molecules at higher concentrations will be a great complement to the single-molecule sensitivity of fluorescence resonance energy transfer (FRET) spectroscopy (30), fluorescence correlation spectroscopy (FCS) (31), two-photon fluorescence microscopy (29), and other fluorescence techniques, because broadband CARS spectra and fluorescence spectra can be obtained simultaneously. This could be useful, for example, in determining how a certain protein (at low concentrations) co-localizes within domains of different lipid composition. Phase changes of lipids and lipid-like molecules also can be studied via both CARS and fluorescence (6,29), and such changes are expected to play a key role in lipid microdomain formation and function (20).
Conclusion
Single-pulse, interferometric CARS microscopy is a sensitive, chemically selective two-dimensional imaging technique. Using a single laser beam significantly simplifies the technique by eliminating many of the instabilities associated with using two separate, synchronized laser beams. By acquiring the broad fingerprint-region vibrational spectrum from 400 cm–1 to 1500 cm–1 obtained at each pixel in the sample, analysis based upon several spectral features can be used to construct detailed images of the sample's chemical composition, even for sample components with peaks at the same frequencies. Three-dimensional imaging also can be performed with the current experimental system by depth profiling the spatial focus. This label-free imaging is an exciting new tool for the direct visualization of segregated chemical domains in polymer photoresists and in biological cells, where direct imaging has proven to be very challenging. While necessary sensitivity improvements are currently underway to achieve chemically selective, label-free, rapid imaging of single lipid bilayers, preliminary results have been extremely promising.
Acknowledgments
The authors gratefully acknowledge the Department of Energy, contract DE-AC02-05CH11231 for funding of the instrumentation and the National Science Foundation for support of personnel. A.G. Caster acknowledges a National Science Foundation Graduate Research Fellowship for support. O. Nicolet acknowledges the support of the Swiss National Science Foundation.
Allison G. Caster and Stephen R. Leone are with the Departments of Chemistry and Physics, University of California, Berkeley, and Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California.
Sang-Hyun Lim is assistant professor with the Department of Chemistry and Biochemistry, University of Texas, Austin.
Olivier Nicolet is a scientific collaborator with The Laboratory of Optoelectronics of Molecular Materials at the Ecole Polytechnique Fédérale de Lausanne, Switzerland.
References
(1) N. Dudovich, D. Oron, and Y. Silberberg, J. Chem. Phys. 118, 9208–9215 (2003).
(2) D. Oron, N. Dudovich, and Y. Silberberg, Phys. Rev. Lett. 89, 273001(1-4) (2002).
(3) D. Oron, N. Dudovich, and Y. Silberberg, Phys. Rev. Lett. 90, 213902(1-4) (2003).
(4) J.X. Cheng, A. Volkmer, L.D. Book, and X.S. Xie, J. Phys. Chem. B 106, 8493–8498 (2002).
(5) C.L. Evans, E.O. Potma, and X.S. Xie, Opt. Lett. 29, 2923–2925 (2004).
(6) M. Müller and J.M. Schins, J. Phys. Chem. B 106, 3715–3723 (2002).
(7) K.P. Knutsen, B.M. Messer, R.M. Onorato, and R.J. Saykally, J. Phys. Chem. B 110, 5854–5864 (2006).
(8) R. Porter, F. Shan, and T. Guo, Rev. Sci. Inst. 76, 043108(5) (2005).
(9) T.W. Kee and M.T. Cicerone, Opt. Lett. 29, 2701–2703 (2004).
(10)T.W. Kee, H. Zhao, and M.T. Cicerone, Opt. Exp.14, 3631–3640 (2006).
(11)H. Kano and H. Hamaguchi, Opt. Exp. 14, 2798–2804 (2006).
(12)A. Zumbusch, G.R. Holtom, and X.S. Xie, Phys. Rev. Lett. 82, 4142–4145 (1999).
(13)J.X. Cheng, L.D. Book, and X.S. Xie, Opt. Lett. 26, 1341–1343 (2001).
(14)E.O. Potma and X.S. Xie, ChemPhysChem 6, 77–79 (2005).
(15)E.O. Potma, X.S. Xie, L. Muntean, J. Preusser, D. Jones, J. Ye, S.R. Leone, W.D Hinsberg, and W. Schade, J. Phys. Chem. B 108, 1296–1301 (2004).
(16)X. Nan, E.O. Potma, and X.S. Xie, Biophys. J. 91, 728–735 (2006).
(17)H. Wang, Y. Fu, P. Zickmund, R. Shi, and J.X. Cheng, Biophys. J. 89, 581–591 (2005).
(18)A. Volkmer, J. Phys. D: Appl. Phys. 38, R59–R81 (2005).
(19)D. Lin-Vien, N.B. Colthup, W.G. Fateley, and J.G. Grasselli, The Handbook of Infrared and Raman Characteristics Frequencies of Organic Molecules (Academic Press, San Diego, 1991).
(20)K. Simons and W.L.C. Vaz, Annu. Rev. Biophys. Biomol. Struct. 33, 269–295 (2004).
(21)S.H. Lim, A.G. Caster, and S.R. Leone, Phys. Rev. A 72, 041803(4) (2005).
(22)S.H. Lim, A.G. Caster, and S.R. Leone, J. Phys. Chem. B 110, 5196–5204 (2006).
(23)L. Lepetit, G. Chériaux, and M. Joffre, J. Opt. Soc. Am. B 12, 2467–2474 (1995).
(24)E.O. Potma, C.L. Evans, and X.S. Xie, Opt. Lett. 31, 241–243 (2006).
(25)W.C. Johnson, J. Wang, and Z. Chen, J. Phys. Chem. B 109, 6280–6286 (2005).
(26)Y.S. Huang, T. Karashima, M. Yamamoto, and H. Hamaguchi, Biochem. 44, 10009–10019 (2005).
(27)J.X. Cheng, Y.K. Jia, G. Zheng, and X.S. Xie, Biophys. J. 83, 502–509 (2002).
(28)Olympus America, Inc. UIS2 Series Biological Microscope Objectives, http://www.olympusamerica.com/seg_section/uis2/seg_uis2.asp. (accessed July 2006).
(29)K. Gaus, E. Gratton, E.P.W. Kable, A.S. Jones, I. Gelissen, L. Kritharides, and W. Jessup, Proc. Nat. Acad. Sci. 100, 15554–15559 (2003).
(30)R.M. Clegg, Curr. Opin. Biotechnol. 6, 103–110 (1995).
(31)K. Bacia, D. Scherfeld, N. Kahya, and P. Schwille, Biophys. J. 87, 1034–1043 (2004).
















AI-Powered SERS Spectroscopy Breakthrough Boosts Safety of Medicinal Food Products
April 16th 2025A new deep learning-enhanced spectroscopic platform—SERSome—developed by researchers in China and Finland, identifies medicinal and edible homologs (MEHs) with 98% accuracy. This innovation could revolutionize safety and quality control in the growing MEH market.


New Raman Spectroscopy Method Enhances Real-Time Monitoring Across Fermentation Processes
April 15th 2025Researchers at Delft University of Technology have developed a novel method using single compound spectra to enhance the transferability and accuracy of Raman spectroscopy models for real-time fermentation monitoring.




