Dealing With the Masses: A Tutorial on Accurate Masses, Mass 32 Uncertainties, and Mass Defects
Spectroscopy
This tutorial illustrates the most important definitions used in high-resolution mass spectrometry and clarifies the misconceptions surrounding some of the relevant terms used in this field.



This tutorial illustrates the most important definitions used in high-resolution mass spectrometry and clarifies the misconceptions surrounding some of the relevant terms used in this field, such as mass resolution, mass accuracy, and mass defect. Examples are given for recent advanced applications of accurate mass measurements, mass defect labels for protein identification, and mass defect filtering techniques in drug metabolism studies.
Today's use of mass spectrometry (MS) is very different from applications just 10 years ago, because the availability of modern hybrid mass analyzers, such as quadrupole-time-of-flight (Q-TOF), quadrupole-Fourier-transform ion cyclotron resonance (FT-ICR), or linear-ion trap-orbitrap instruments, has led to a multitude of selective scan functions, data processing routines, and structural identification tools. Mass-based structural identification of unknown compounds such as drug metabolites from complex biological matrices often involves high-resolution MS measurements because tandem mass spectrometry (MS-MS) spectra from regular triple-quadrupole or ion-trap instruments usually do not confirm unambiguously the identity of a compound in a sample. A precise, high-resolution measurement of the accurate mass of the molecule, on the other hand, can provide the empirical formula of that compound, aiding the structural identification process tremendously. In these experiments, it is important to measure the mass-to-charge ratio (m/z) with the smallest mass uncertainty possible. Ideally, the analytical protocol provides sufficient resolution and mass accuracy to yield a single empirical formula for every compound analyzed by comparing the experimental results to the calculated exact masses. Of course, the situation becomes even more complicated when isobaric interferences are present because overlapping signals lead to shifts in the measured m/z values. Except for isomeric ions, isobaric signals in mass spectra will always lead to inhomogeneous peak broadening in the spectra because of the different mass defects of the elements, if the mass spectrometer has an insufficiently high resolving power. On the other hand, knowing the mass defect of expected compounds in a complex sample can be used to simplify the obtained results by using so-called mass defect filters.
This last paragraph is probably confusing for beginners to MS, but the use of as many of the relevant terms common in high-resolution MS as possible was intentional. The underlying principles and theories are not trivial and often not fully understood, even by more experienced mass spectrometrists. For example, while measurements of high mass accuracy (i.e., how close is the experimentally determined accurate mass to the calculated exact mass) are performed almost always on instruments with high resolving power, it is sometimes possible to obtain very accurate mass measurements from low-resolution instruments. Moreover, high mass accuracy is available only from properly mass-calibrated instruments, while resolving very close species in a spectrum only requires sufficient resolving power. Simply stated, the use of high resolution alone does not guarantee accurate results. To correctly interpret the results from such experiments, it is vital to have a keen understanding of the theory and instrumentation involved in the process.
It is the goal of the present article to provide a basic introduction to the most important definitions and principles used in high-resolution MS and to clarify some of the misconceptions surrounding these terms. Further, we will explain briefly the most common mass analyzers used for high-resolution mass measurements and describe some recent advanced applications of accurate mass measurements, such as mass defect labels for protein identification and mass defect filtering techniques used in drug metabolism studies.
Some Basic Definitions
The unit of measurement used in MS is the unified atomic mass unit (u); defined as 1/12 the mass of a 12C atom (1 u equals 1.66054029 × 10–27 kg). In other words, the mass of a 12C atom is exactly 12 u. This selection is simply a convention and, interestingly, before 1961, 16O was used for the same purpose. Two important terms are nominal mass and monoisotopic mass. The nominal mass of a molecule is defined as the sum of the integer masses of the most abundant isotopes in a molecule. For example, let us consider molecular nitrogen, N2, and ethene, C2H4. These molecules have different empirical formulae; however, their associated nominal masses are the same: N2, 2 × 14 = 28 u is equal to C2H4, 2 × 12 + 4 × 1 = 28 u. Mass spectrometers with insufficient mass resolving power such as quadrupole or ion-trap MS will not be able to distinguish these two molecules after ionization. The two resulting ions also are said to be isobaric ions.
The monoisotopic mass of a molecule differs from the nominal mass in that the sum of the exact masses of the most abundant (not the lightest!) isotopes of the constituent atoms is used instead of the integer masses. The exact mass of an atom is the result of the masses of its nucleons, while taking into account its nuclear binding energy (this will be addressed in detail in the next section). In the literature, the term exact mass is used more commonly instead of monoisotopic mass. For the earlier example, the calculated monoisotopic (exact) masses are 2 × 14.00307 = 28.00614 and 2 × 12 + 4 × 1.007825 = 28.03130 for N2 and C2H4, respectively. It is clear that while these molecules have the same nominal mass, they have different monoisotopic masses. It follows that, due to the noninteger monoisotopic masses, different empirical formulas will never result in the same monoisotopic mass.
Finally, the average mass includes the weighted average of all the naturally occurring isotopes of the constituting elements of the molecule. For larger molecules, such as peptides and proteins, the average mass can be significantly larger than the monoisotopic mass. Importantly, the monoisotopic mass is not necessarily the most abundant peak in the mass spectrum.
The Mass Defect
The mass defect is simply the difference between the exact mass and the nominal mass. This defect is characteristic for every atom. For example, the 16O atom is composed of eight protons, eight neutrons, and eight electrons with individual masses of 1.007276470, 1.008664904, and 0.000548579903 u, respectively, adding up to a total mass of 16.131919633 u, which is larger than the monoisotopic value of 15.994915 u. To explain this mass defect, we must look at the nuclear binding energy. Binding energy is required to separate the protons and neutrons in the atomic nucleus, and the observed mass defect arises from the relativistic loss of mass occurring when this binding energy is released during the formation of a stable atomic nucleus. Figure 1 illustrates the binding energy and the associated mass defect for the first 90 elements of the periodic table.


Figure 1
The two forces acting on the protons and neutrons that compose the nucleus are the attractive nuclear force and coulombic repulsion. These forces act in unison and generate the resulting binding energy and observed mass defect. The nuclear force is a relatively strong attractive force that acts to bind together the protons and neutrons to form a stable atomic nucleus. Although it is a strong attractive force, the nuclear force has an effective range of only a few proton radii. Opposing the nuclear force is coulombic repulsion, existing between the positively charged protons. The coulombic force is substantially weaker than the nuclear force; however, it is effective over a much longer range. Coulombic repulsion increases quadratically with the addition of protons compared with the stronger nuclear force that increases linearly with increasing numbers of protons and neutrons. This interplay of forces allows the elements to be placed into two broad categories, as observed in Figure 1. Light elements with atomic numbers of less than 56 (iron) have nuclei in which the nuclear force is dominant, allowing for the formation of stable atoms (large binding energy). In this region, there is a rapid increase in the binding energy with each addition of protons and neutrons to form larger elements. Elements with atomic numbers greater than 56 have sufficient numbers of protons in their associated nuclei so that the repulsive coulombic force begins to destabilize the nucleus, in turn lowering the overall stability (binding energy) as the atomic mass increases.
From Figure 1, we observe that the first five elements before 12C in the periodic table exhibit positive mass defects compared with the negative defects seen for larger atoms. This is simply the result of the selection of 12C as the reference point for the unified atomic mass unit. As we will see in a later section, this characteristic mass defect can be used as a means of identifying and characterizing specific molecules in complex samples.
A slight variation of the mass defect definition leads to the Kendrick mass defect. The Kendrick mass scale was first developed in 1963 as a way of simplifying the analysis of complex mixtures of organic substances containing compounds with extensive methylene (CH2) repetitions (1). To reduce the complexity of these data sets, a mass scale based upon CH2 rather than 12C was implemented, where CH2 is defined as exactly 14 u as opposed to 14.01565 u in the 12C scale. The Kendrick mass of a molecule is then calculated as follows:
Kendrick mass = 12C mass × 14/14.01565.
The Kendrick mass defect is the difference between the exact and the nominal Kendrick mass. The prime advantage of the Kendrick mass scale is that members of a homologous series differing only in the degree of alkylation will all exhibit the same mass defect. A plot of Kendrick mass defect versus nominal Kendrick mass greatly simplifies complex MS data by providing information about the class of compounds from the y axis and the number of CH2 units from the x axis. Marshall and coworkers have demonstrated a number of interesting applications of this technique in combination with high resolution FT-ICR data (Figure 2).



Figure 2
Accurately Measuring Masses and Determining Elemental Formulae
The term mass accuracy refers to how close the mass measured by the mass spectrometer comes to the calculated exact mass of an ion. Importantly, if the mass measurement can be made with sufficient accuracy, a unique empirical formula can be assigned. The number of possible empirical formulae calculated from the data decreases rapidly with increasing mass accuracy (Figure 3). In the absence of a unique empirical formula, analysis can be highly ambiguous. Often, the experimentally determined accurate mass measurements do not exhibit the required measurement certainty needed for unambiguous elemental formula assignment within a window of accepted mass uncertainties. In these cases, a series of potential molecular formulae are possible and it is usually left to experienced operators, or software programs, to select the most likely candidate by applying additional means of identification, such as isotope ratios.



Figure 3
The mass accuracy of a measurement is reported commonly as a relative mass error or mass uncertainty (parts per million), as shown in the following equation:


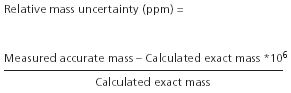
Unique elemental formulae often can be found for very small molecules with moderate mass measurement uncertainties because the relative error is mass dependent. This is illustrated by considering our now-familiar example of N2 and C2H4, differing by only 0.02516 u or 25.16 millimass units (mmu) between their calculated exact masses. At the nominal mass of m/z 28 and considering C, H, N, and O as possible atoms in the positive ion mode, no species come within 400 ppm of each other (N2 versus CO), and there is almost 900 ppm between N2 and C2H4. The problem is, of course, that the required mass accuracy increases exponentially with increasing mass, as seen in Figure 3.
Resolution and Resolving Power
Accurate mass measurements are performed most often using high-resolution instruments. High-resolution experiments do not, however, automatically guarantee accurate results. First of all, the use of high-resolution instruments can ensure that the peak associated with a particular m/z value is free of other interfering species, which could otherwise lead to peak shifts and inaccurate results. Importantly, accurate m/z measurement capability is possible only if the mass analyzer can resolve adjacent peaks in very complex samples. Figure 4 illustrates an example in which an FT-ICR instrument was able to resolve four adjacent species in an MS-MS spectrum. Sometimes even low-resolution mass spectrometers can be used for this purpose. Naturally, for an instrument to generate accurate mass data, the instrument must be properly mass-calibrated.



Figure 4
The ability of an instrument to separate closely spaced peaks is termed resolving power. Resolution is calculated from the acquired data and is used to quantify the separation between peaks in a mass spectrum. In other words, an instrument can be said to have high resolving power, whereas the resulting spectra are said to be of high resolution.
There are two common methods for determining the resolution in a spectrum. The first method is used for spectra generated on magnetic sector instruments: the 10% valley definition. In this definition, two peaks of equal intensity are said to be resolved when the overlapping region between the peaks is equal to 10% or less of the intensity of the original peaks. The resolution R is given by the following equation:


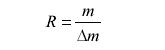
where m is the mass of the heavier peak and Δm is the mass difference between the two species. The second method, now routinely applied, uses the full width at half maximum (FWHM) of the peak. For FWHM calculations, resolution is reported as m/z divided by the width of the peak at half its height. It is important to realize that for a given spectrum, the two definitions do not give the same values. For perfectly Gaussian peaks, FWHM values are 1.8× larger than those obtained from the 10% valley definition.
The criteria for whether spectra are considered low- or high-resolution are not defined clearly. Generally, low-resolution spectra exhibit R < 2000, and high-resolution spectra show R > 5000. Of course, there is a trade-off between resolution and sensitivity, and the desired resolution usually is the minimum required to solve the specific problem under investigation. Modern high-resolution instruments can produce spectra with resolutions in the tens of thousands and in some cases, as we will see in the next section, the hundred thousands to million regions.
Instruments for Accurate Mass Measurements
This section briefly summarizes the most important mass analyzers for high resolution and accurate mass measurements, namely, magnetic sector, time-of-flight (TOF), Fourier transform-ion cyclotron resonance (FT-ICR), and orbitrap instruments. The interested reader is referred to textbooks on mass spectrometry such as Gross's excellent Mass Spectrometry (2) for a more detailed description of the instruments.
In the past, high-resolution experiments were carried out using double-focusing magnetic sector instruments, consisting of an electrostatic analyzer and a magnetic sector. The two sectors focus ions based upon different principles. The double-focusing instrument consists of an electrostatic analyzer operating as a kinetic energy separator coupled to momentum separation carried out via the magnetic sector. These instrument designs still are quite useful in specialized applications, and resolving powers >70,000 are possible with modern double-focusing machines with mass uncertainties between 1 and 5 ppm (3).
The TOF mass analyzer is the most straightforward design among the instruments discussed here. This analyzer takes advantage of the fact that ions of different mass (m/z) will take differing times to traverse a certain distance, given that they both experience an equal initial accelerating force. Light ions will reach the detector first, with heavier ions requiring longer periods to travel the same distance. Modern TOF instruments usually are equipped with a reflectron to enhance resolution. Also, the TOF analyzer is often the final stage in the common hybrid design of Q-TOF instruments. Resolving powers of up to 20,000 have been demonstrated, and the mass accuracies can be within less than 5 ppm with appropriate mass calibration (2).
The ion cyclotron resonance mass analyzer is based upon the trapping of ions using both magnetic and electrostatic fields. In FT-ICR mass spectrometry, a trapping cell is placed in the center of a spatially uniform magnetic field B. This magnetic field is provided by a superconducting magnet similar to those used in nuclear magnetic resonance (NMR) spectroscopy. Ions are trapped by combined magnetic and electrostatic fields, which cause the ions to undergo cyclotron motion characteristic of their m/z value. The frequency of this cyclotron motion is measured and converted from the time domain to the frequency domain using the Fourier transform. Very high resolving powers have been reported (up to 3,300,000) with mass errors of often less than 1 ppm (4).
The orbitrap is a relatively new instrument, and its operation is somewhat similar to that of the quadrupole ion trap. The orbitrap makes use of a static electrostatic field to trap ions, however, as opposed to the dynamic radio frequency field used in quadrupole ion traps. The orbitrap consists of a two-electrode system composed of a thin wire inner electrode and an electrically isolated coaxial outer electrode. A static electromagnetic field is generated, which traps the ions radially in stable trajectories. The motion of trapped ions can be described as a simple harmonic oscillator with a characteristic frequency that is dependent upon the m/z value of the ion. As with FT-ICR, the resulting time domain data are converted to a frequency domain spectrum using a Fourier transformation. Resolutions of up to 150,000 have been reported along with sub-part-per-million relative mass uncertainties (5).
Although mass analyzers for accurate mass measurements are usually high-resolution instruments, sometimes low-resolution mass spectrometers can be used for measuring m/z values with high accuracies (6–8). Several studies have demonstrated relative mass uncertainties of <10 ppm for molecules with m/z between 200 and 6000 on triple-quadrupole and ion-trap instruments. Of course, because one never knows in advance whether potential interferences are present in unknown samples, only high mass resolving power can give sample-independent, reliable mass measurements.
Some Interesting Applications Using Mass Defect
Mass Defect Labeling for Peptides and Proteins
Most peptides and proteins exhibit similar mass defect characteristics because amino acids contain principally only five elements: C, H, O, N, and S. Their mass defects are 0, 0.0078, -0.0051, 0.0031, and -0.0279, respectively, generating a net mass defect for a protein molecule. Of course, carbon does not affect the mass defect of a protein, and the combined mass defects of oxygen and nitrogen tend to cancel each other, resulting in virtually no net mass defect shifts. Furthermore, because sulfur is not very abundant in proteins, it does not influence the total mass defect greatly. As a result, hydrogen is the dominating element in the mass defect for the entire protein molecule. Proteins can have large mass defects, increasing by approximately 0.05 u for every 100 u increase in mass. Amster and coworkers have shown that despite these large mass defects, the distribution of masses is quite narrow (Figure 5). This example demonstrates that peptides are clustered tightly within each individual unit mass value. Because of the large amount of overlap within these clusters, the ability to unambiguously identify a peptide from accurate mass data alone is very difficult.



Figure 5
Several methods have been developed to incorporate mass defect labels into either peptides or proteins, with the goal of altering the mass defect of a peptide through added groups that exhibit significant mass defects. The incorporated label will shift the mass to unpopulated regions of the unit mass scale, thereby reducing the complexity of these regions. This shift will decrease possible interferences and enhance the number of identified proteins.
In the following section, we discuss two interesting examples for mass defect labels. The first involves bromine for shifting the mass defect of either peptides or intact proteins. In the second example, rare earth metals are utilized.
Amster and colleagues developed a method for the identification of peptides generated from a tryptic digest of whole cell lysates of M. maripaludis. In their experiments, the side chain of cysteine was derivatized with 2,4-dibromo-(2'-iodo) acetanilide. Figure 6a shows the distribution of the relative mass defects for all tryptic peptides. Figure 6b demonstrates the effect of cysteine labeling, with singly labeled cysteine-containing peptides centered on the left of the main distribution and the doubly labeled species on the right. This shift to unpopulated regions of the unit mass scale increases the number of identified derivatized peptides compared with underivatized peptides and should increase the number of identified proteins. To demonstrate the effectiveness of their technique, the authors examined the M. maripaludis proteome using matrix-assisted laser desorption–ionization (MALDI) FT-ICR MS, which enabled them to identify 304 proteins in a cysteine-labeled sample compared with 268 proteins in an unlabeled sample.



Figure 6
A similar technique was developed by Schneider and colleagues (9,10). Their technique was to label intact proteins with a bromine-tag by coupling the protein N-terminus with 3-bromo-1-(5-carboxy-pentyl)-pyridium bromide-NHS ester (BDOPP). This method differed from the previous method in that tandem MS experiments on the labeled proteins were used for protein identification. The technique was used for differential expression experiments, in which the ability to detect whether a protein in a sample is up- or down-regulated is probed through the use of isotopically pure "light" and "heavy" labels. In this case, the light label includes a 79Br atom and the heavy analog has 81Br. As with the previous method, proteins were detected after imparting a characteristic mass defect that aids in identification and helps to reduce chemical interferences.
Finally, Meares and colleagues developed a label for cysteine side chains, utilizing bromoacetamidobenzyl-1,4,7,10-tetraazacyclododecane-N,N',N'',N'''-tetra-acetic acid ligands bound to one of three possible rare earth metals: terbium (Tb), yttrium (Y), and lutetium (Lu) (Figure 7). After derivatization, the labeled peptides were first bound to an antibody column, allowing the removal of interferences from unlabeled species. After elution, the labeled peptides were submitted to MS analysis. As with the previous methods, the rare earth element labels shifted the mass defect of the peptides by a characteristic amount. Because three different metals were available, the resulting mass defect could be fine-tuned depending upon the complexity of the biological samples.


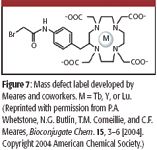
Figure 7
Mass Defect Filters for Metabolite Identification
The characteristic mass defects of biological molecules also can be used to simplify complex MS data from biological samples, which often contain abundant, interfering isobaric peaks. The general idea is to remove interfering signals of compounds for which the mass defects fall outside of an operator-determined range. This relatively new filtering technique was developed by Zhang and colleagues (11) and has been applied successfully in drug discovery applications for metabolite identification. In these applications, a postacquisition mass defect filter is applied to high-resolution liquid chromatography (LC)–MS data of metabolite samples. After filtering the data, the accurate masses of metabolites can be determined and empirical formulae are calculated, followed by tandem MS experiments to further elucidate the metabolite structures. Importantly, the filter is applied to the raw LC–MS data and simply points to the location of potential metabolites in the chromatograms, leaving the original acquisition data unchanged. Essentially, it is a visualization tool that works by removing chemical noise from the spectra. A number of different mass defect filters can be applied, without the need for time-consuming data re-acquisitions.
Drug metabolites generally can be assigned to two categories: those with nominal mass and mass defect similar to the parent drug molecule and those with significantly different values. These two categories require different filter templates.
The first group of metabolites, with similar nominal mass and mass defect as compared to the parent drug, are screened with a so-called drug filter template. Good examples are metabolites from monohydroxylation of the parent drug molecule. Hydroxylation adds 16 u to the nominal mass and shifts the mass defect by a characteristic –0.0051 u. In this case, setting the filter with a nominal mass range of ±20 u centered on the protonated parent drug molecule and the mass defect range to ±0.010 u will reveal possible metabolite peaks for monohydroxylation reactions.
The second group of metabolites exhibits significantly different nominal masses and mass defects in comparison to the parent drug. In these cases, a core structure template is used for screening, taking into account only a characteristic portion of the parent drug instead of the entire molecule. This template filter is used for metabolites formed as a result of a drastic modification of the parent drug. Consider the case in which the parent drug compound is cleaved in half, forming two smaller subunits. These subunits will have significantly smaller nominal masses than the parent drug and may or may not have a similar mass defect. The use of this drug template filter would not detect these metabolites because they fall outside the typical mass ranges around the parent.
An illustrative example is shown in Figure 8, where a mass defect filter was applied to raw data from an LC-electrospray-QTOF instrument for identifying metabolites of the drug omeprazole in human plasma. The unprocessed data in Figure 8a exhibit a significant abundance of interfering species, obscuring the presence of metabolites. The same data after processing via the appropriate mass defect filters, on the other hand, clearly illustrate the location of metabolites in the chromatograms.


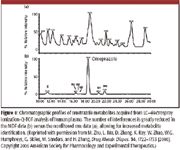
Figure 8
Summary
As a result of the elemental mass defects, all chemical compounds of the same nominal molecular mass have a different calculated exact mass and elemental formula, respectively, except for isomers. The accurate measurement of m/z values therefore allows the determination of the elemental formula for an unknown compound in a sample, if the precision of the experimental measurement is sufficiently high. Usually, high-resolution mass spectrometers, such as magnetic sector, Q-TOF, FT-ICR, or orbitrap instruments, are implemented for these accurate mass measurements. On the other hand, the different mass defects also will lead to inhomogeneous peak broadening in the spectra if interfering isobaric species are present in the sample. In these cases, the mass measurements can give inaccurate results if the employed mass spectrometer does not provide adequate resolving power.
The mass defects in protein and peptide identification are dominated largely by hydrogen, with very narrow distribution of masses within each nominal mass value. By incorporating appropriate chemical mass defect tags, it is possible to shift the peptide–protein mass to unused areas in this nominal mass space, thereby increasing the number of identified peptides and proteins. The second example for utilizing the mass defect is the mass defect filter. This filter is used to simplify raw mass spectral data from biological samples, usually for the purpose of drug metabolite identification. This visualization tool works by removing interfering signals of compounds for which the mass defects fall outside of an operator-determined range.
Dr. Dietrich Volmer is Head of Bioanalytical Sciences at the Medical Research Council's Collaborative Centre for Human Nutrition Research in Cambridge, UK. His main research interests are in different areas of biological mass spectrometry. Dr. Volmer is also a faculty member in the Department of Chemistry at Dalhousie University in Halifax, Nova Scotia.
Andrew Leslie is a graduate student in Dr. Volmer's group at Dalhousie University in Halifax.
References
(1) E. Kendrick, Anal. Chem. 35, 2146–2154 (1963).
(2) J.H. Gross, Mass Spectrometry: A Textbook (Springer-Verlag, Berlin, Heidelberg 2004).
(3) D.H. Russell and R.D. Edmondson, J. Mass Spectrom. 32, 263–276 (1997).
(4) F. He, C.L. Hendrickson, and A.G. Marshall, Anal. Chem. 73, 647–650 (2001).
(5) Q. Hu, R.J. Noll, H. Li, A. Makarov, M. Hardman, and G. Cooks, J. Mass Spectrom. 40, 430–443 (2005).
(6) G.P. Paul, W. Winnik, N. Hughes, H. Schweingruber, R. Heller, and A. Schoen, Rapid Commun. Mass Spectrom. 17, 561–568 (2003).
(7) O. Fiehn, J. Kopka, R.N. Trethewey, and L. Willmitzer, Anal. Chem. 72, 3573–3580 (2003).
(8) A.N. Tyler, E. Clayton, and B.N. Green, Anal. Chem. 68, 3561–3569 (1996).
(9) L.V. Schneider and M.P. Hall, Drug Discov. Today 10, 353–363 (2005).
(10) M.P. Hall, S. Ashrafi, I. Obegi, R. Petesch, and J.N. Peterson, J. Mass Spectrom. 38, 809–816 (2003).
(11) H. Zhang, D. Zhang, and K. Ray, J. Mass Spectrom. 38, 1110–1112 (2003).














High-Speed Laser MS for Precise, Prep-Free Environmental Particle Tracking
April 21st 2025Scientists at Oak Ridge National Laboratory have demonstrated that a fast, laser-based mass spectrometry method—LA-ICP-TOF-MS—can accurately detect and identify airborne environmental particles, including toxic metal particles like ruthenium, without the need for complex sample preparation. The work offers a breakthrough in rapid, high-resolution analysis of environmental pollutants.


The Fundamental Role of Advanced Hyphenated Techniques in Lithium-Ion Battery Research
December 4th 2024Spectroscopy spoke with Uwe Karst, a full professor at the University of Münster in the Institute of Inorganic and Analytical Chemistry, to discuss his research on hyphenated analytical techniques in battery research.

Mass Spectrometry for Forensic Analysis: An Interview with Glen Jackson
November 27th 2024As part of “The Future of Forensic Analysis” content series, Spectroscopy sat down with Glen P. Jackson of West Virginia University to talk about the historical development of mass spectrometry in forensic analysis.

Detecting Cancer Biomarkers in Canines: An Interview with Landulfo Silveira Jr.
November 5th 2024Spectroscopy sat down with Landulfo Silveira Jr. of Universidade Anhembi Morumbi-UAM and Center for Innovation, Technology and Education-CITÉ (São Paulo, Brazil) to talk about his team’s latest research using Raman spectroscopy to detect biomarkers of cancer in canine sera.


