Quantitative Mass Spectrometry: Part I
In this month's installment, columnist Ken Busch discusses how to best answer the questions "What is it?" and "How much is there?"



We leave the interpersonal dynamics of laboratory analysis, and the persistent temptation to request informal data and transform those answers into confirmed results, for another column, which will appear in this space in March 2085. The scenario lampooned above has existed since mass spectrometry (MS) first became a common analytical tool in the laboratory. Determining the identity of a sample, or characterizing the components of a mixture, often is thought of as the bread and butter of MS. Providing a quantitative measure is more difficult for a number of reasons, many of which are related to the fact that the time and resources needed to develop and confirm a valid analytical procedure are invariably in short supply. Quantitation with MS can provide extraordinarily precise and accurate results. In specialized applications, such as the long-term monitoring of dioxin residues in human fat, the achievements are simply phenomenal. But the time and resources invested to achieve such a result are also significant.



Kenneth L. Busch
In the absence of a rigidly constrained protocol, such as that required in a new drug application or other highly regulated analysis, time and resources to support the development and completion of a quantitation will be limited. Even a brief review of the literature, especially in reports that appear outside of a peer-review process, provides many examples of "almost right" quantitation. The answers might not be "too far wrong," nor are they expected to be. It is simply that the quality of the analytical results is concordant with the effort invested. The numerical answer might be close to correct, but the analyst cannot certify that it is.
Despite the long history of quantitative MS, the topic usually is afforded only a limited discussion in current specialized texts. Perhaps this is because the methods of quantitation are considered to be covered adequately in general analytical texts used in the undergraduate curriculum. Indeed, there is usually a broad overview of the use of calibration curves, or the method of standard additions, and a discussion of internal versus external standards, in such texts. We understood those concepts just fine, theoretically, in class. It was only when we were put into the laboratory and forced to generate such analytical data ourselves that the difficulty of the task became apparent. It took a great deal of time, and in light of a full curriculum, we soon moved on to some other topic and some other laboratory exercise. The important central concepts of how a calibration curve, or a method of standard addition, is used in an MS quantitative analysis return here as the core of this series of columns. This author, at least, is many decades removed from the sweat and effort of that first laboratory quantitation. (Not that I remember the grade of B that I received. Not at all.) The more removed we are from the basics, and the need for specific attention to details, the more likely our errors become. The result is the "almost right" quantitation. It is useful to review what we must do to provide better quantitative results. Millard's excellent reference work (1) has been supplemented recently by a text by Lavagnini and colleagues (2). Detailed discussions of quantitative practices in MS appear in the current specialized literature, from which exemplars will be taken and discussed in this series.
In this first column of this five-part series, we first disconnect quantitation from the mass spectrum itself, emphasize some very basic assumptions that underlie quantitation, and review the role of internal and external standards. In the second column, we will review the possible influence of matrix effects, sample preparation errors, the need for proper experimental design, and an overview of basis statistics. In the third column, we will discuss the use of the calibration curve, with details specific to its use in MS, and statistics associated with its creation, evaluation, and presentation. In the fourth column, we will describe similar attributes of the standard addition method. In these columns, we use examples taken from the published literature. In the fifth and final column of this series, we will discuss the use of isotopically labeled internal standards. The ability to use such standards is unique to MS. However, choosing this approach engenders significant costs associated with preparation of and certification of these standards. Rather than talk in the abstract, we will use quantitative analysis using gas chromatography–mass spectrometry (GC–MS) or liquid chromatography–mass spectrometry (LC–MS) as examples. Special protocols can be used to quantify samples without sample separation. These methods are encountered more rarely, but they will be previewed in the final column in the series.
One of the key points taught in the short course "Introduction to Mass Spectrometry" is that the mass spectrum of a particular compound is independent of the amount of compound introduced into the ion source. This statement is usually true across a dynamic range of at least a few orders of magnitude. At higher amounts of sample, and higher instantaneous concentration of sample in the source, the appearance of the mass spectrum can change, usually due to the occurrence of ion–molecule reactions. At lower amounts of sample, signals from the sample might become difficult to distinguish from chemical noise in the mass spectrum. These matrix effects are different from the consequence of a changing flux of sample into the ion source (as in the elution of a sample from a gas chromatograph), which in conjunction with scanning of a mass analyzer can lead to changes in the apparent relative abundances of the ions in the mass spectrum. For the latter, the analyst averages mass spectra recorded across both the rising and the falling portions of an eluted GC peak to create a mass spectrum similar to what would be measured if the sample concentration were constant. For the former matrix effects, "averaging" of spectral data is not useful at higher concentrations, but can be useful in distinguishing signal from noise at lower concentrations.
Relevant to the topic at hand, the usefulness of mass spectral libraries in identification of compounds depends upon the fact that the mass spectrum reflects the ion structure and unimolecular dissociation chemistry (in electron ionization), and is independent of the amount of compound present. In the introductory short course, I have learned that relentless attention to basics avoids confusion later on. Accordingly, we do so here. A classic mass spectrum appears with units of m/z on the x axis, and the term "relative abundance" on the y axis. The term "relative intensity" also is used sometimes, and while the difference between abundance and intensity is real, the key point here is the adjective "relative." The most intense (most abundant) peak in the mass spectrum is assigned a value of 100, and all of the other peaks are plotted in the mass spectrum (or tabulated) relative to that value. The actual measured intensity of that base peak could be 4230, 76,531, or 230,654 units. The other peaks in the mass spectrum scale proportionately. A beginning student makes a natural connection between the height of the peak on the y axis and the "response" of the mass spectrometer to be used in quantitation. The disconnect between "relative intensity" in a mass spectrum and absolute signal intensity must be emphasized strongly.
It is the absolute measured intensity of ion signal that is proportional to the amount of sample in the ion source, and the absolute intensity that ultimately will be used as part of quantitation. To derive a quantitative measure, we must connect via proportionality the intensity measured for the unknown sample to the intensity measured for some standard of known amount. That proportionality is apparent in a calibration curve graph, or in the intercept in a method of standard addition. However, we begin with the even more basic assumptions that underlie quantitation. Among the implicit assumptions is the fact that the identity of the compound to be quantitated is known; the targeted compound is available as a pure certified standard; there is no (or a known) matrix effect in the response of the compound; and variation in instrument performance is a known or a negligible contributor to overall error. Quantitation becomes more difficult, and the accuracy of quantitative data obtained decreases, when these assumptions are not true.
Sample identity. It seems self-evident that the identity of the compound to be quantitated would be known. Usually it is. However, structural isomers and stereoisomers are different compounds, with sometimes significantly different chemical behaviors and analytical responses, and the analyst should always take the time to consider all of the potential confounding variables, just as in the use of homologous or analogous compounds as standards.
Purity of standard. Because quantitation with MS will involve at some point a proportional response between the sample (unknown) and a standard (known), the purity of the standard must be assessed. The assessment must include such factors as stability of the standard as well as lot-to-lot synthetic variations. The stability of the prepared standard solution also must be assessed. Because the purity of the standard itself usually is assessed via MS, such consideration begets a propagation of errors analysis.
Matrix effects. A propagation of errors analysis properly evaluates the relative contributions of sources of error in each independent variable to the error in the final result. In quantitation with MS, the potential matrix effects are both variable and ill defined, and therefore difficult to include in the propagation of errors analysis. The errors that accrue from simplifying or removing the matrix, with sample cleanup, sample derivatization, chromatographic separation, and the like are almost always less than the errors that arise because we cannot predict the variable effect of the matrix. This is exactly why we carry out these processes, and this is why we often use internal standards introduced as early in the sample processing stream as possible. In so doing, we might deduce an estimate of sample recovery as well as error. Recovery is usually less than 100%. There are methods (radioactive tracing is one) that trace losses at each step of a process and provide a relative scale of the matrix effect. Without the time to pursue these studies, we (amazingly) simply accept less than 100% recoveries in the analytical process.
Instrument performance. Quantitative MS, as a topic, also includes procedures for calibration of the mass scale of an instrument, but these will not be dealt with in this series. In quantitation of an unknown, we often blithely assume that instrument performance is constant and does not affect the accuracy or precision of our measurement significantly. Millard (1) lists relevant instrument performance variables in an electron ionization source as mass measurement accuracy, filament emission variability, electron energy variation, and repeller voltage variation. A list also can be compiled for factors relevant to other ionization methods, and to the performance of the mass analyzer, and (less commonly now) changes in the responsiveness of the detection and amplification and data recording system of the instrument.


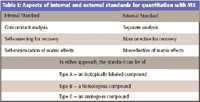
Table I: Aspects of internal and external standards for quantitation with MS
Finally, as we finish this introductory column, we review the basic concepts of the use of internal and external standards in a quantitation using MS. Lavagnini and colleagues (2) delineate a useful scheme for the overall design of a quantitative experiment in which the selection of either an internal or an external standard plays an early pivotal role, and in which the cost–benefit analysis also is introduced as a variable. (In the evaluation of the cost and benefits, the "almost right" analytical result alluded to earlier can become a "good enough" result.) The difference between an internal standard and an external standard, and all of the variations in each method, comes down to one simple difference, summarized in Table I. The external standard method always implies an analysis separated in time and space from the analysis of the sample itself. In such an analysis, response variations due to matrix effects, sample recovery, and a host of other experimental factors cannot be included in the propagation of errors analysis. In an internal standards method, the standard (in whatever form) is added to the sample as early in the overall process as practicable. The purpose of doing so is to ensure that whatever happens to the unknown target happens to the known standard as well, with the ideal hope that any errors will (within the final proportionality comparison) "cancel out." Deviation from this ideal has been discussed within the analytical community for decades, and the discussion is not reproduced here. Although the use of the internal standard usually is held to be preferable, it is not always so. Suffice it to say that, based upon the availability of time and resources, excellent results can be obtained with either the internal or the external standard method. In the next column, we will discuss the use of proper experimental design an dappropriate statistics in quantitative mass spectrometry.
Kenneth L. Busch reveals that the third question often asked of mass spectrometrists by folks who provide samples is "Can you do it right away?" Of course, a quick turnaround on any one sample results in a parallel expectation for all subsequent samples. After all, every task is easy as long as it is someone else doing the work. This column has no connection to the National Science Foundation. The author can be reached at WyvernAssoc@yahoo.com.
References
(1) B.J. Millard, Quantitative Mass Spectrometry (Heyden, Philadelphia, 1978).
(2) I. Lavagnini, F. Magno, R. Seraglia, and P. Traldi, Quantitative Applications of Mass Spectrometry (John Wiley and Sons, New York, 2006).

















AI-Powered SERS Spectroscopy Breakthrough Boosts Safety of Medicinal Food Products
April 16th 2025A new deep learning-enhanced spectroscopic platform—SERSome—developed by researchers in China and Finland, identifies medicinal and edible homologs (MEHs) with 98% accuracy. This innovation could revolutionize safety and quality control in the growing MEH market.





New Raman Spectroscopy Method Enhances Real-Time Monitoring Across Fermentation Processes
April 15th 2025Researchers at Delft University of Technology have developed a novel method using single compound spectra to enhance the transferability and accuracy of Raman spectroscopy models for real-time fermentation monitoring.

