Calibration: Effects on Accuracy and Detection Limits in Atomic Spectroscopy
Whenever concentrations are measured with atomic spectroscopy, they are calculated from a calibration curve. Therefore, attaining accurate quantitative results requires proper calibration, especially when measuring low-level concentrations near detection limits. This brief tutorial explains the effect of calibration on the accuracy and detection limits in atomic spectroscopy analyses. A thorough discussion of statistics in analytical chemistry is available in a series of 52 articles (1), with six articles (2–7) being the most relevant to this discussion.
Understanding Calibration and Linearity
Linearity must be considered when performing analyses by any atomic spectroscopy technique. In general, the linearity of atomic absorption (AA) is ≈3 orders of magnitude, inductively coupled plasma–optical emission spectrometry (ICP-OES) is ≈6 orders of magnitude, and inductively coupled plasma–mass spectrometry (ICP-MS) is ≈10–11 orders of magnitude. Although this is technically true, it does not mean that calibrating over wide ranges will produce accurate results. If a calibration curve in ICP-MS is constructed from calibration standards over 9 orders of magnitude (that is, 1 ppt to 1000 ppm) and has a correlation coefficient of 0.999992, this would be considered a linear calibration curve with an excellent correlation coefficient, implying that any concentration between 1 ppt and 1000 ppm would be read accurately against this curve, which is not true.
Theoretical Example
Consider a theoretical example of a calibration curve made from standards ranging from 0.1 to 100 ppb—four orders of magnitude. The only way to measure accurate concentrations over a wide range is if every measurement is perfect, which is not possible: every standard on the curve has an error associated with it. Consider that if each calibration standard in Figure 1 has a 2% error on the intensity, the mean intensity error increases as the concentration and intensity of each standard increases: a 2% error on the 0.1 ppb standard (100 counts per second [cps]) is only ±2 cps, but a 2% error on the 100 ppb standard (95,000 cps) is ±1,900 cps. If these data are plotted and a regression (or best-fit) line drawn through the standards (Figure 1), the error of the higher concentration standards dominates the curve. The best-fit line passes almost directly through the 100 ppb standard, but the lower standards are increasingly farther away from the line, demonstrating that the highest absolute error contributes most to the overall curve fit.


FIGURE 1: Calibration curve with 2% error bars for each standard.
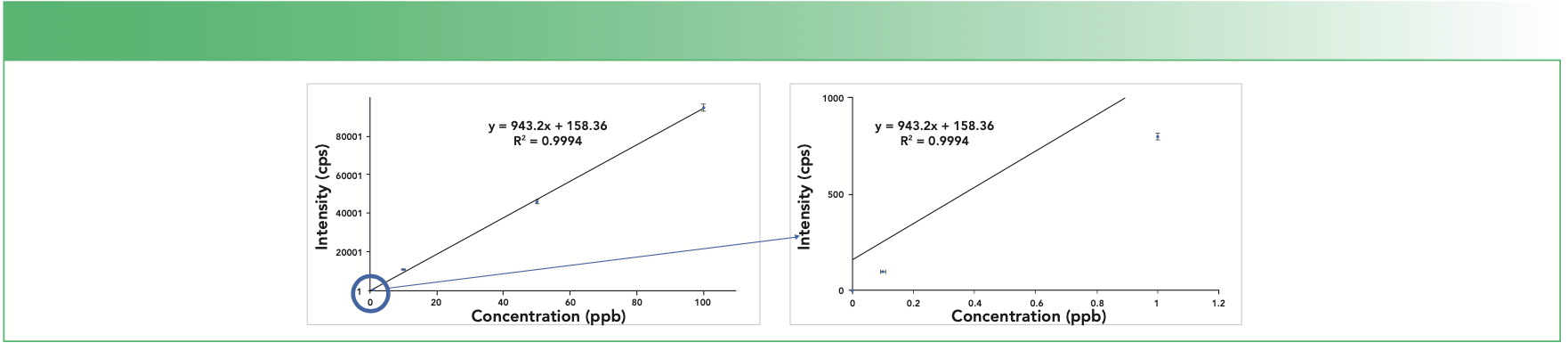
Since the error of high concentration standards dominates the calibration curve, both accuracy at lower concentrations and detection limit calculations are affected. If accuracy at low concentrations is the most important criteria, then the calibration curve should be constructed without the use of high-concentration calibration standards: the calibration curve should only contain low-level standards. For example, if selenium (Se) is measured by ICP-MS and is expected to be below 10 ppb in most samples with a reporting limit of 0.1 ppb, a calibration curve consisting of a blank and three standards at 0.5, 2.0, and 10.0 ppb will provide much better accuracy at levels of 0.1 ppb than a calibration curve with standards at 0.1, 10, and 100 ppb. This same principle also applies to ICP-OES and AA, even though their linear ranges are narrower than ICP-MS.
ICP-MS Example
Now consider an example from ICP-MS: a calibration curve for Zn constructed from 11 standards (0.01, 0.05. 0.1, 0.5, 1 5, 10 50, 100, 500, and 1000 ppb) over six orders of magnitude. Figure 2a shows the resulting calibration curve, which has a correlation coefficient (R2) of 0.999905, indicating excellent linearity. However, when the 0.1 ppb standard was analyzed as a sample against this curve, it read 4.002 ppb – a huge error.

 ICP-MS calibration curve for Zn over six orders of magnitude with 11 standards; (b) same curve, with the lowest seven standards expanded.")
FIGURE 2: (a) ICP-MS calibration curve for Zn over six orders of magnitude with 11 standards; (b) same curve, with the lowest seven standards expanded.
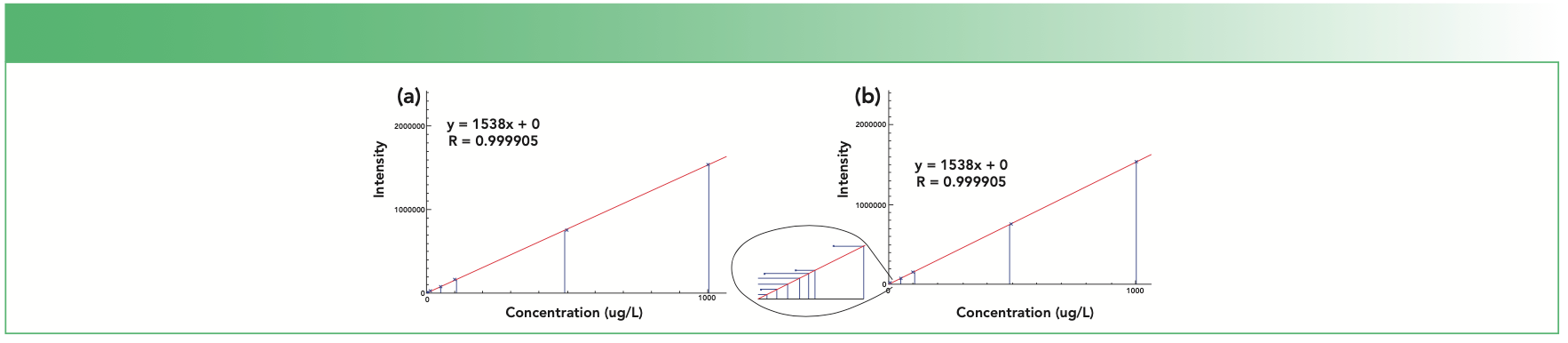
The cause of this high readback is apparent in Figure 2b, which shows an expanded view of the low end of the curve (the 0.01–10 ppb standards): Zn contamination in the seven lowest standards which causes them to read higher than their nominal concentrations. However, this issue is not apparent from the correlation coefficient since the lowest standards contribute almost nothing statistically to the curve, compared to the four highest standards. This example clearly demonstrates the problem of calibrating at concentrations much higher than expected in samples.
The Effect of Contamination
The previous example demonstrates the problem with contamination in calibration standards, but another common issue is contamination in the calibration blank. Contamination can originate from a number of sources, including reagents (acid, water), deposition in the sample introduction system, or deposition in the instrument itself (interface cones, for example), to name a few. Since the blank is assumed to have analyte concentrations of zero, the measured signal from the blank is subtracted from all subsequent measurements. If the blank signal intensity is higher than that of a standard (or sample), the blank-subtracted concentrations will be negative, resulting in poor calibration curves. However, this may not be apparent with calibration curves which include high-concentration standards because the contamination will be small relative to the signal from the high standards. The result: the correlation coefficient will indicate a linear regression, implying that accurate results can be obtained at low concentrations.
In reality, there is no such thing as a true “blank”: contamination always exists at some level. The goal is to limit contamination so that it is much lower than the lowest calibration standard.
Calibrating for Low and High Concentrations Levels
As shown, accurate results for low-level samples can best be obtained by calibrating at concentrations close to the expected concentrations. But this leads to the question: will low-level calibrations produce accurate results for samples with high concentrations? The answer is yes, in most cases. The reason: the error at high concentrations dominates the curve. This means that errors at low concentrations will not have as large an impact on high-concentration samples. To confirm, it is good practice to perform a linear range study on any set of calibration standards: run successively higher standards against the calibration curve. The linear range is generally considered the highest concentration that recovers within 10% of its true value.
However, there are situations where high concentrations will be negatively affected by low level concentrations. Two common causes are large errors in the low calibration standards or when the analyte concentrations are so high that they cause matrix suppression. However, this latter case can usually be compensated for by proper selection of an internal standard.
Summary
The key to accurate measurements at low concentrations and to achieving meaningful detection limits is to establish calibration curves with low-level standards. Just about any calibration curve can produce correlation coefficients when using high-concentration standards, but these will not provide meaningful, accurate low-level results or detection limits.
References
(1) D. Coleman and L. Vanatta, Am. Lab. Parts 1–52, (2002–2013). https://www.americanlaboratory.com/841-Search/?search=Coleman%20Vanatta%20Part%201
(2) D. Coleman and L. Vanatta, Am. Lab. Part 40 (2010). https://www.americanlaboratory.com/914-Application-Notes/1114-Part-40-Blanks/
(3) D. Coleman and L. Vanatta, Am. Lab. Part 41 (2011). https://www.americanlaboratory.com/914-Application-Notes/981-Statistics-in-Analytical-Chemistry-Part-41-Blanks-Concluded/
(4) D. Coleman and L. Vanatta, Am. Lab. Part 44 (2011). https://www.americanlaboratory.com/913-Technical-Articles/35232-Part-44-R-sup-2-sup/
(5) D. Coleman and L. Vanatta, Am. Lab. Part 45 (2011). https://www.americanlaboratory.com/913-Technical-Articles/35860-Part-45-R-sup-2-sup-Continued/
(6) D. Coleman and L. Vanatta, Am. Lab. Part 46 (2012). https://www.americanlaboratory.com/913-Technical-Articles/38042-Part-46-R-Concluded/
(7) D. Coleman and L. Vanatta, Am. Lab. Part 47 (2012). https://www.americanlaboratory.com/913-Technical-Articles/39480-Part-47-Risky-Calibration-Practices/
Kenneth Neubauer is a Principal Application Scientist with PerkinElmer in Shelton, Connecticut. Direct correspondence to: kenneth.neubauer@perkinelmer.com ●



 Measurements")

LIBS Illuminates the Hidden Health Risks of Indoor Welding and Soldering
April 23rd 2025A new dual-spectroscopy approach reveals real-time pollution threats in indoor workspaces. Chinese researchers have pioneered the use of laser-induced breakdown spectroscopy (LIBS) and aerosol mass spectrometry to uncover and monitor harmful heavy metal and dust emissions from soldering and welding in real-time. These complementary tools offer a fast, accurate means to evaluate air quality threats in industrial and indoor environments—where people spend most of their time.





Atomic Perspectives: Highlights from Recent Columns
March 3rd 2025“Atomic Perspectives,” provides tutorials and updates on new analytical atomic spectroscopy techniques in a broad range of applications, including environmental analysis, food and beverage analysis, and space exploration, to name a few. Here, we present a compilation of some of the most popular columns.

