An Enhanced Immunoaffinity Enrichment Method for Mass Spectrometry–Based Translational Proteomics
Translational proteomics has not been as successful as originally anticipated. Because mass spectrometry (MS) can separate proteins at the sequence level, it provides the selectivity needed for this application; however, traditional challenges still exist, including time-to-result, throughput, and sample-size requirements.
Translational proteomics has not been as successful as originally anticipated. Because mass spectrometry (MS) can separate proteins at the sequence level, it provides the selectivity needed for this application; however, traditional challenges still exist, including time-to-result, throughput, and sample-size requirements. For analytical validation and verification purposes, sample preparation times must be reduced from days to hours. Scientists recently coupled a previously developed immunoaffinity enrichment method to selected reaction monitoring (SRM) MS. The MS immunoassay–SRM method combines liquid chromatography–tandem mass spectrometry (LC–MS-MS) for target identification, a microscale immunoaffinity capture method for enrichment, and subsequent SRM analysis. Using the MS immunoassay–SRM workflow, a standard high-throughput method for developing targeted biomarker identification of proteins in human plasma and serum for clinical research was developed.
Mass spectrometry (MS)-driven proteomics has made progress in the identification and quantification of disease biomarkers, including C-reactive protein as an indicator for myocardial infarction and prostate-specific antigen (PSA) for prostate cancer. Despite these and other successes, translational proteomics, which is defined as the translation of biomarker discovery to routine analysis, has not been nearly as successful as originally anticipated because comprehensive proteomic analysis of plasma, serum, and other biological fluids has proved exceedingly challenging. The "look alike" nature of molecular isoforms, the enormous dynamic range of protein concentrations of potential interest (>10 orders of magnitude in blood plasma), and the fact that molecules of interest are often in low abundance have all slowed the progress of biomarker hunters across the globe.
One example of the challenge presented by protein analyte isoforms is demonstrated by the need to distinguish between full-length parathyroid hormone (PTH) 1-84 and multiple N-terminally truncated PTH variants. The differences between these isoforms are critical to accurate diagnosis of endocrine and osteological diseases (1). Similarly, in clinical testing, PSA typically presents in truncated and modified isoforms, making precise detection and quantification difficult which contributes to a high false-positive rate (2).
From a processing point of view, binding protein detachment and the high dynamic range of samples have hampered assays for insulin-like growth factor 1 (IGF-1), a marker for growth-related illness that is important in cell proliferation, differentiation, apoptosis, and tissue growth from a research point of view (3).
To add to these challenges, verification and population-scale biomarker validation require the analysis of hundreds or even thousands of high-quality samples. Sample collection and storage must use standard protocols to reduce potential variations attributable to endogenous enzymes or sample contamination. Verification and validation studies require multiple control groups and subjects in disease subcategories — all gathered over the course of disease progression. The analysis of many samples is required to distinguish normal human genetic heterogeneity and heterogeneity attributable to disease. High-throughput detection methods are essential to achieving statistical confidence in the accurate identification of molecules of interest (4).
Challenges of Current Assay Technologies
The current gold standard for biomarker detection uses enzymes to bind antibody ligands to target molecules. The enzyme-linked immunosorbent assay (ELISA) has played a central role in diagnostic and clinical research applications for the last two decades. However, as molecular biology advances and as scientists are able to access new technologies with higher recognition accuracy, ELISA is emerging as a potential source of inaccuracy leading to imprecise biomarker detection and resulting in incorrect diagnosis and treatment. In certain cases, demonstrated poor concordance among assays from different manufacturers has raised questions about ELISA's accuracy (5). Poor concordance stems from the variety of proprietary antibodies used to detect different epitopes, as well as natural biological diversity manifested in post-translational modifications, single nucleotide polymorphisms, and cross-reactivities between antibodies to off-target proteins. Various interferences have also been observed, including antireagent antibodies and endogenous auto-antibodies, which can generate incorrect results. These indirect, nonspecific signals mask consequential variations in molecular structures (5). An October 2013 study investigated the specificity of the commercial CUZD1 ELISA assay (6). CUZD1 is a biomarker for ovarian and pancreatic cancer, as well as inflammatory bowel disease (7). Using a combination of western blot, high performance liquid chromatography (HPLC), and MS, the study confirmed that instead of CUZD1, the commercial assay recognizes a nonhomologous cancer antigen, CA125. The study concluded that the poor characterization of the commercial ELISA assays may lead to false biomarker discovery (6).
A Combined Effort
MS has been used for quantification of small molecules for years and, more recently, has been applied to protein quantification and analysis. Because MS can separate proteins at the sequence level, it provides the selectivity needed to address the specificity issues outlined above (4). As noted, for validation and verification purposes, high throughput and high accuracy are vital; unfortunately, the traditional challenges surrounding MS-based techniques include time-to-result, throughput, and sample-size requirements. For MS to be used for validation and verification purposes in translational proteomics, sample preparation times must be reduced from days to hours (4). Despite their shortcomings, both ELISA and MS methods are widely used in routine clinical research and analysis. In recent years, researchers have used immunoaffinity enrichment coupled to MS to combine the selectivity of immunoaffinity extraction with the specificity of MS detection (8,9).
Sample Processing Before MS
One of the hurdles that scientists have worked to overcome to improve MS accuracy involves sample preparation before MS. The wide dynamic range of proteins in biological fluid samples compromises the ability of MS to achieve sufficient sensitivity to accurately quantify potential biomarkers. Scientists have explored numerous sample preparation approaches to eliminate proteins of little interest that increase dynamic range. These approaches include fractionation using multiple liquid chromatography (LC) columns, depletion of abundant proteins, enrichment using solid-phase extraction, nanoparticles, and immunoaffinity enrichment by various techniques that include magnetic beads and microcolumns (10).
Method
Scientists at Thermo Fisher Scientific, in partnership with Arizona State University, the Institut de Recherche Clinique de Montréal, the Sahlgrenska Academy at the University of Gothenberg, the University Health Network at the University of Toronto, and Kings College Hospital in London, have coupled a previously developed immunoaffinity enrichment method (MSIA, Thermo Fisher Scientific) (8) to selected reaction monitoring (SRM) MS (11). The MS immunoassay–SRM approach combines LC–MS-MS for target identification, a microscale immunoaffinity capture method for enrichment, and subsequent SRM analysis.
Target Identification
To identify optimal peptides and transitions for SRM, the system combines algorithmic and spectral library prediction using Pinpoint software with results from high-resolution LC–MS-MS analysis resulting in a list of targets. After the first strategy is built and recombinant protein or peptide standards are obtained, scientists can narrow the list by iteratively optimizing collision energies, LC gradient, and SRM scheduling windows. Only the highest intensity transitions are retained and, after three or four repetitions, the system is ready for sample analysis. Figure 1 shows the workflow for the optimization of SRM analyses.


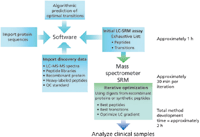
Figure 1: Automated workflow for optimization of SRM analyses.
Enrichment
In the immunoaffinity capture step, antibodies are surface-immobilized in small, porous microcolumns fixed into pipette tips. Samples are repeatedly aspirated and dispensed through these affinity pipettes to expose the immobilized antibody to the targeted proteins (10). This iterative step has the equivalent effect of lengthening an LC column. After the proteins are captured, they are eluted for subsequent MS detection. In a single iterative separation step, target proteins and variants are captured using antibodies aimed at epitopes in the protein sequence. The approach is applicable to low-abundance proteins because of the robust immunoaffinity enrichment of the target analyte. Figure 2 summarizes the immunoaffinity enrichment workflow.


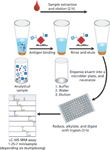
Figure 2: Immunoaffinity enrichment workflow.
Reference Standards
Most targeted protein detection methods add surrogate peptides and corresponding isotope-labeled counterparts to samples for standardization after digestion, just before the MS step. The MS immunoassay–SRM design, however, adds internal reference standards at the outset, before enrichment. This means the standard goes through all the same steps that the sample undergoes, acting as a normalizer for all processing and data collection steps. In this type of MS immunoassay–SRM experimental workflow, recombinant proteins rather than isotope-labeled peptides are used. Recombinant proteins, if available, are less expensive and can be easier to obtain. After digestion, heavy isotope-labeled peptides are added to ensure quality control of the LC and MS detection steps in each sample to measure run-to-run variance.
Results
Using the MS immunoassay–SRM workflow, a standard high-throughput workflow for developing targeted biomarker identification of proteins in human plasma and serum was developed (11). The standardized protein identification workflow included targeted methods for 16 different protein analytes, including members of the apolipoprotein family: ApoE, ApoA1, ApoCI, ApoCIII, and ApoJ; some medium-high abundance proteins: ceruloplasmin, vitamin D binding protein, beta-2 microglobulin, and C-reactive protein; and several important low-abundance proteins, including procalcitonin, PTH, IGF-1, PSA, erythropoietin, proprotein convertase subtilisin/kexin type 9, and amyloid beta.
Results for PTH and IGF-1 MS immunoassay–SRM were compared to clinical analyzer immunoassays. Correlation between the two methods yielded R2 values ranging from 0.67 to 0.87, depending on the surrogate target peptide. For PTH, the correlation of the analyzer data with MS immunoassay–SRM of the single, N-terminal peptide versus the average of all peptides suggests that the analyzer-based immunoassay likely includes uncharacterized, intact, and truncated protein species, leading to an overestimation of functional protein. In comparison, SRM characterizes all isoforms; therefore, all species of interest are individually quantified, resulting in improved accuracy. In addition, the method allowed discovery of previously undescribed isoform variants of PTH.



Table I: Assay format comparison: MS immunoassay tip versus beads
Subsequently, Thermo Scientific scientist Eric Niederkofler presented data in a webinar (12) that compared IGF-1 results obtained using the MS immunoassay–SRM approach with results obtained using a magnetic bead immunoenrichment technique. This work demonstrated that the MS immunoassay technique was five times more effective than the bead method, lowering the limit of quantification by 10-fold (see Table I) and producing improved signal-to-noise ratio at the MS stage (Figure 3).



Figure 3: High-resolution orbital trap MS-MS data of IGF-1 peptide obtained from MS immunoassay tips (top) versus magnetic beads (bottom).
In our published results (11), we presented linear regression curves that showed R2 values from 0.089 to 0.098. In Figure 4 we present similar data for two apolipoprotein E peptides.


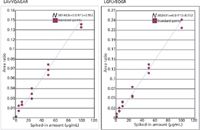
Figure 4: Calibration curves for ApoE targeted peptides.
Sensitivity and Precision
Figure 5 presents SRM chromatograms for ApoE surrogate peptides from digested human plasma. Peak shapes are symmetrical and relatively free of significant noise. SRM transition ion ratios were within ±15% of internal standard reference ratios. To ensure selectivity, we used several SRM transitions for each peptide. Detection limits and dynamic ranges for all targeted peptides were well within the useful range for the selected analytes, and lower limit of quantification values were all within the lower portion of existing ranges.


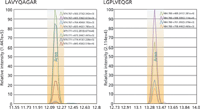
Figure 5: Representative SRM chromatogram results.
Observations
Protein or Peptide Level Enrichment?
While developing this approach to immunoaffinity enrichment over the last five years, we found that, in many cases and for several reasons, antiprotein antibodies are preferable binding agents to antipeptide antibodies. Antiprotein antibodies typically enrich multiple protein isoforms, making it easier for SRM analyses to distinguish active from inactive forms. Because antipeptide antibodies must be highly specific, each target requires a separate antibody. For example, for PTH protein detection using MS immunoassay–SRM and antipeptide antibodies, investigators would need to generate seven different antipeptide antibodies to reveal the same information available from one antiprotein antibody. Using an antiprotein antibody raised toward an epitope common to all isoforms or variants costs less, because only one protein is required to bind multiple forms. Using antipeptide antibodies also makes it more difficult to add new targets to a test, because multiple new antipeptide antibodies must be developed for each analyte.
Antiprotein antibody capture provides distinct advantages in the trypsin digestion stage as well. Because digestion depends on the protein-to-trypsin ratio, complex samples such as raw plasma or serum require relatively large amounts of trypsin and long digestion times. Samples enriched at the protein level are digested after enrichment. Because they are less complex than raw samples, this results in reduced digestion times, typically 3 h or less.
In comparison, samples enriched at the peptide level require crude digestion before enrichment and, as a result, take more time to process. Crude digestion also injects a potential source of error stemming from uncontrolled or unknown causes. By their nature, samples from different states of disease are expected to contain varying levels of proteins. Digestion variability because of illness may impact results and has not been adequately studied. Finally, if surrogate peptides are not unique, digestion and subsequent enrichment may result in incorrect measurements, because one peptide sequence may be present in multiple proteins.
Enrichment for All
In our observation, all samples can benefit from immunoenrichment, not just those in which tests are searching for low-abundance molecules. Enrichment of high-abundance targets reduces processing times, lowers costs, and improves specificity. First, the lower the sample complexity, the faster it can go through trypsin digestion. Second, the process uses less trypsin than if the sample were unenriched; using less trypsin makes a test less expensive. Third, although protein targets may be in high abundance, important isoforms are probably rarer, and enrichment promotes isoform capture. The work with PTH and ApoAI showed that immunoenrichment and detection with the MS immunoassay–SRM method identified new isoforms, as well as new C- and N-terminally truncated variants, which may be present in much lower amounts. A practical advantage lies in reduced cleaning and maintenance requirements for mass spectrometers and LC columns because they are not injected with plasma or serum. Using purified samples also improves routine measurement consistency.
Multiplexing
Multiplexed ELISA or other immunoassay technologies present limitations because as the number of analytes increases, sensitivity typically decreases. The immunoenrichment technique allows multiplexing of analytes by serial extraction of the same sample. To test for different targets, samples can be enriched using different antibodies embedded in the microcolumns on the pipette tips. This approach is fast, conserves sample volumes, and avoids the problem of accommodating several antibodies with different optima for binding and elution conditions on a single substrate.
Conclusion: Immunoenrichment for MS-Based Translational Proteomics
MS-based proteomics is a powerful tool for research and facilitates two important areas: identification, verification, and validation of new biomarkers, and detection and quantification of those markers in clinical research settings. Potential clinical research and biopharmaceutical applications might include screening, tracking progression, predicting course and outcome, monitoring for recurrence, and personalized assessment of drug response and toxicity (4). MS immunoassay–SRM–based proteomics, using the immunoenrichment method described, may offer a significantly shorter development time, reduced costs, and higher specificity for the identification of proteins and their isoforms in comparison to existing methods.
Mary Lopez and Bryan Krastins are with the Thermo Fisher Biomarkers Research Initiatives in Mass Spectrometry Center (BRIMS) in Cambridge, Massachusetts. Direct correspondence to: mary.lopez@thermofisher.com
References
(1) M. Lopez et al., Clin. Chem. 56(2), 281–290 (2010).
(2) H.B. Carter, Asian J. Androl. 14, 355–360 (2012).
(3) E. Niederkofler et al., PLoS ONE 8(11), e81125 (2013).
(4) E.S. Baker et al., Genome Med. 4, 63 (2012).
(5) J. Becker and A. Hoofnagle, Bioanalysis 4(3), 281–290 (2012).
(6) I. Prassas et al., Clin. Chem. 60(2), 290–291 (2014).
(7) C. Liaskos et al., Clin. Dev. Immunol., vol. 2013, article ID 968041 (2013). Available at: http://dx.doi.org/10.1155/2013/968041.
(8) R. Nelson and C.R. Borges, J. Am. Soc. Mass Spectrom. 22(6), 960–968 (2011).
(9) O. Stoevesandt and M. Taussig, Expert Reviews Proteomics 9(4), 401–414 (2012).
(10) D. Nedelkov, Expert Rev. Mol. Diagn. 12, 235–239 (2012).
(11) B. Krastins et al., Clin. Biochem. 46, 399–410 (2013).
(12) E. Niederkofler, "MSIA-SRM: Next Generation Method for Insulin-Like Growth Factor-1 Measurement," (2012). Available at: http://www.thermoscientific.com/en/about-us/events/Solve-Challenging-Protein-Quantitation-Problems-in-Clinical-Research-with-Mass-Spectrometric-Immunoassay-Application-MSIA.html#sthash.2AhLquTf.dpuf.











High-Speed Laser MS for Precise, Prep-Free Environmental Particle Tracking
April 21st 2025Scientists at Oak Ridge National Laboratory have demonstrated that a fast, laser-based mass spectrometry method—LA-ICP-TOF-MS—can accurately detect and identify airborne environmental particles, including toxic metal particles like ruthenium, without the need for complex sample preparation. The work offers a breakthrough in rapid, high-resolution analysis of environmental pollutants.


The Fundamental Role of Advanced Hyphenated Techniques in Lithium-Ion Battery Research
December 4th 2024Spectroscopy spoke with Uwe Karst, a full professor at the University of Münster in the Institute of Inorganic and Analytical Chemistry, to discuss his research on hyphenated analytical techniques in battery research.

Mass Spectrometry for Forensic Analysis: An Interview with Glen Jackson
November 27th 2024As part of “The Future of Forensic Analysis” content series, Spectroscopy sat down with Glen P. Jackson of West Virginia University to talk about the historical development of mass spectrometry in forensic analysis.

Detecting Cancer Biomarkers in Canines: An Interview with Landulfo Silveira Jr.
November 5th 2024Spectroscopy sat down with Landulfo Silveira Jr. of Universidade Anhembi Morumbi-UAM and Center for Innovation, Technology and Education-CITÉ (São Paulo, Brazil) to talk about his team’s latest research using Raman spectroscopy to detect biomarkers of cancer in canine sera.


