GLP and GMP Approaches to Method Validation — Going the Same Way?
A review of the United States Pharmacopeial Convention (USP) stimulus paper on the revision process to adopt quality-by-design principles for the development, validation, and operation of analytical procedures used for good manufacturing practice (GMP) analysis - and how it compares to a draft guidances by the Food and Drug Administration (FDA) for industry on validation of analytical and bioanalytical procedures.



In September 2013, the United States Pharmacopeial Convention (USP) published a stimulus to the revision process to adopt quality-by-design principles for the development, validation, and operation of analytical procedures used for good manufacturing practice (GMP) analysis. In February 2014, this was followed by a draft Food and Drug Administration (FDA) guidance for industry on validation of analytical procedures. Also, the FDA issued a new draft that updates their 2001 guidance for industry on bioanalytical method validation. Are the approaches the same? What do you think?
I live in London and we have a saying here: You wait a long time for a bus and then three turn up at once. Recently, in method validation a similar thing has occurred and three draft documents for method validation have been issued over a relatively short time span. Two of these method validation documents are intended for laboratories following good manufacturing practice (GMP) principles and one is for bioanalytical laboratories following good laboratory practice (GLP) principles.
The three documents are
- G.P. Martin and colleagues, "Lifecycle Management of Analytical Procedures: Method Development, Procedure Performance Qualification, and Procedure Performance Verification," published in Pharmacopeial Forum in September 2013 (1);
- "Analytical Procedures and Methods Validation for Drugs and Biologics," draft Food and Drug Administration (FDA) guidance for industry issued in February 2014 (2); and
- "Bioanalytical Method Validation," draft FDA guidance for industry dated September 2013 focused on GLP (3).
We devote this column to reviewing the contents of each of these documents, and comparing and contrasting their approaches to a critical subject for quantitative analysis: the validation of analytical methods or procedures. I will spend most of the column looking at the United States Pharmacopeial Convention (USP) proposal for method development and validation of analytical procedures (1) because this is the most encompassing and radical of the three approaches. Some of these publications refer to analytical procedures and analytical methods — from my perspective the terms are equivalent, and I will use them interchangeably.
Analytical method development, procedure validation, and transfer to other laboratories to carry out routine analysis are key parts of the traditional analytical process. This starts, if you're lucky, with defining what you want the method to do and ends up with the operational use of the procedure. In the middle is the stuff that should be done properly, but usually is not, as evidenced by how poorly a method operates in routine use. This oversight typically occurs because the method development and validation is performed by one group and the operation of the procedure is carried out by another group. In many instances, the two groups rarely talked to each other except when things went wrong — and blamed each other for the problems. Of course, this never happens in your organization, does it?
In the Beginning . . .
Method validation is not new because there have been a number approaches over the years in both GMP and GLP quality disciplines. Validation of GMP analytical procedures was harmonized with the agreement of the International Conference on Harmonisation (ICH) quality publications Q2A and Q2B covering the text and methodology of validation of analytical procedures respectively (4,5) in 1994 and 1996, respectively. These two publications were merged into a single document with the revision as ICH Q2(R1) in 2005 without changing any text (6). The first bioanalytical methods validation conference was held in 1990 (I was a co-chair of one of the sessions), sponsored by the FDA and the American Association of Pharmaceutical Scientists (AAPS), that drew up the first guidelines for bioequivalence and bioavailability studies typically carried out under GLP (7). This led to the FDA issuing a guidance for industry on bioanalytical methods validation in 2001 (8).
How Robust Is Your Method?
Validation of an analytical method or procedure, regardless of the GLP or GMP, measures parameters such as precision, accuracy, measurement uncertainty, and limits of quantification. One parameter that I want to focus on is robustness. This is defined in ICH Q2(R1) (6) as follows:
The robustness of an analytical procedure is a measure of its capacity to remain unaffected by small, but deliberate variations in method parameters and provides an indication of its reliability during normal usage.
In the methodology section, there is more information on the topic of robustness (6):
The evaluation of robustness should be considered during the development phase and depends on the type of procedure under study. It should show the reliability of an analysis with respect to deliberate variations in method parameters.
If measurements are susceptible to variations in analytical conditions, the analytical conditions should be suitably controlled or a precautionary statement should be included in the procedure. One consequence of the evaluation of robustness should be that a series of system suitability parameters (e.g., resolution test) is established to ensure that the validity of the analytical procedure is maintained whenever used.
This is the interesting bit — for a document focused on the validation of analytical procedures we have the statement that robustness is actually a job to be done during method development. How many laboratories actually conduct robustness studies? How many laboratories actually conduct robustness studies when under time pressure? However, ICH Q2(R1) does say that robustness depends on the method, but then it goes on to mention some typical variables that could be considered for liquid chromatography (LC) procedures such as variations of pH of the aqueous buffer of a mobile phase, variations in mobile-phase composition, or different columns (different lots or suppliers). Therefore, in my view, robustness should be undertaken during the method development phase. There is no discussion in any of the current pharmacopeia general chapters (9–11) or the bioanalytical guidance dealing with validation on method development (8). However, without developing a robust method and knowing how changing a key parameter will impact a method's performance, the work done during method validation could be, and often is, wasted.
Guidance Versus Regulation
The aim of ICH Q2(R1) (6) was to provide guidance in the validation of any analytical procedure, but this document has tended to take on the role of a regulation. Don't think, just do it. ICH Q2(R1) has been incorporated into the pharmacopeias; for example, USP <1225> on validation of analytical procedures (10). Note the chapter numbering: It is an informational (strong guidance) chapter but not a mandatory general chapter (12).
In the current version of the U.S. Pharmacopeia National Formulary (USP–NF), there are three general chapters involved with analytical procedures:
- USP <1224> on transfer of analytical procedures (9)
- USP <1225> on validation of analytical procedures — this is essentially ICH Q2(R1) (10)
- USP <1226> on verification of compendial procedures (11)
Interestingly, there is no mandatory chapter on the validation of analytical procedures only guidance. The aim of all three chapters is to allow an analytical procedure to become operational in a regulated laboratory as shown in Figure 1. However, there is no consideration of the importance of method development, except where discussed in ICH Q2(R1) as mentioned above. As you can see, method development remained outside of the control of pharmacopoeias — despite its importance to the operational method.


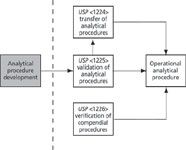
Figure 1: Current USPâNF general chapters on analytical procedures.
ICH Q10, Q9, and Q8: Pharma Is Changing
Since the publication of ICH Q2(R1), the International Conference on Harmonisation has issued three key documents that impact the way that the pharmaceutical industry operates. It changes the industry from reactive to proactive — or should if these documents are implemented correctly. The three documents are
- ICH Q10 on pharmaceutical quality systems (13): This document describes a quality management system based on ISO 9001, but adapted for the pharmaceutical industry. A key change is the introduction of continual (not continuous as pharma is a regulated industry) improvement. In response to the publication of ICH Q10, the European Commission Health and Consumers Directorate-General (EU GMP) chapters 1 (14) and 7 (15) have been issued to incorporate this new approach.
- ICH Q9 on quality risk management (16): This document covers an overall approach to the assessment and management of risk in the pharmaceutical industry.
- ICH Q8(R2) on pharmaceutical development (17): This document is important for the remainder of our discussion because it provides the framework for the approaches that the USP has taken for analytical procedure development, validation, and application.
To understand the approach taken in ICH Q8(R2) we have to step back in time and outside the pharmaceutical industry.
Quality by Design
A catchy phrase is it not? However, this is key to understanding the overall approach proposed by the USP paper. Therefore, we need to consider the meaning of quality by design (QbD). This concept was outlined first by Joseph Juran, a well-known expert and consultant in quality who stated that "product features and failure rates are largely determined in planning for quality" (18). Put simply, quality must be designed into a product or a process, but quality cannot be tested into it. This is the reason why method development is probably more important than method validation — method validation merely confirms that the development was done correctly.
Therefore, without adequate quality steps designed into the procedure during the development phase, such as understanding how an analytical procedure works and identifying the major variables, there is not much point in putting it into operation because it would not be very robust or effective. The ineffectiveness would typically be evidenced in the operational use of the method with more out-of-specification (OOS) results compared to a similar procedure in which time and effort has been taken to understand how any analytical procedure method works. This applies in any GMP or GLP laboratory — you reap what you sow.
Back to the Future
Some of the key terms and definitions from ICH Q8(R2) are presented in Table I (16). Although ICH Q8(R2) is focused on the development of a pharmaceutical product, there are several topics that are pertinent to the development, validation, and operation of an analytical method such as a life cycle, design of experiments, and robustness that form the basis of the USP proposed approach. This is why it's important to understand the role and terminology of ICH Q8(R2). We will meet the modified versions of these terms for analytical procedures when we look at the new USP approach.



Table I: ICH Q8 key terms and definitions
Enter Stage Left: The USP Stimulus Paper
In the September 2013 issue of Pharmacopeial Forum (1), a USP expert committee published a stimulus to the revision process paper entitled "Lifecycle Management of Analytical Procedures: Method Development, Procedure Performance Qualification, and Procedure Performance Verification." This paper proposes a QbD approach to method development, validation, and operational application of an analytical procedure via a single life-cycle concept. The importance of this stimulus paper is that for the first time there would be a formal link between method development and method validation within a pharmacopeia. Just from the title of the paper we can see the adaptation of two ICH Q8(R2) terms: life-cycle management (Q8: life cycle) and procedure performance verification (Q8: continuous process verification). However, there are other terms that are strange, such as procedure performance qualification — also, where is method validation in the title?
Rewriting USP General Chapters
In the stimulus to the revision process paper (1), it is proposed that the three existing USP general chapters dealing with analytical procedure validation, verification, and transfer will be replaced by two general chapters. There will be a mandatory general chapter number <220> dealing with the minimum requirements for method validation and an informational general chapter number <1220> covering the whole life cycle of analytical procedure development, validation, as well as use and modification best practices.
Overview of the Analytical Procedure Life Cycle
Following from the principle of ICH Q8(R2), analytical procedure development, validation, and use and modification is a life-cycle process with defined activities and deliverables. A breakdown of the three-stage process is shown in Figure 2.


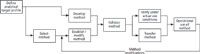
Figure 2: Proposed stages in the analytical procedure life cycle. Adapted from reference 1.
The first stage of the process is method development and understanding. The last word is important! This goes back to Juran (18): Quality is designed into an analytical procedure during the method development stage and is not tested during the validation part of the life cycle. Therefore, you need to understand how the method works by knowing the key variables and how these influence the analysis: The KISS principle applies here. (KISS? Keep it simple, stupid.) Don't make an analytical method more complex than it needs to be.
The second stage is procedure performance qualification — eh? This is method validation under another name and has been taken from the FDA's updated "Guidance for Industry" on process validation (19). In essence, this stage demonstrates that the procedure or a modified procedure is fit for the intended purpose of the assay.
The third stage is the procedure performance verification, which involves checking how the method operates in actual use and that it remains under control. This can be achieved through routine monitoring and charting performance using either Shewart or Cusum plots and reviewing variations in the procedure's performance. If data indicate that the method is not functioning as expected, it should be investigated to see the cause of the variation. Outcomes of an investigation may be a change to the method, which we will discuss later in this column.
Method Development Stage in More Detail
I want to look in more detail at the proposals for the method development stage because I want to discuss the QbD principles that have been incorporated in the overall approach. Within this stage, there are a number of activities as outlined in the USP stimulus paper (1), as explained below.
Define the Analytical Target Profile for the Procedure
The analytical target profile (ATP) is the interpretation of the "Quality Target Product Profile" under ICH Q8(R2) for quality control and analytical development laboratories. An example of an ATP can be measuring analyte X in a specified matrix over the concentration range between A% and B% with a predefined uncertainty for the reportable result.
The ATP may also define any limits of detection if measuring impurities and in the presence of any potentially interfering compounds. In a move away from the typical ICH Q2 definitions, the USP stimulus paper talks about uncertainties not precision and accuracy potentially bringing the pharmaceutical industry into the ISO world. Defining the ATP is an important first step because it is the specification for the method against which the procedure will be validated. Note that the ATP does not mention the instrumental technique used in the method, it merely defines the required performance of the method. So, the first stage of QbD is to put a marker in the sand with the specification for the analytical procedure.
Select the Analytical Technique
Typically, an ATP would not define the analytical technique; thus, in the next stage, the responsible analyst should select an appropriate technique that is able to meet the ATP. In doing so, the ATP will be decomposed into more-detailed critical performance characteristics, which should also include the sampling and a sample preparation element of any analysis because the samples may be liquid, semisolid, or solid. This philosophical approach will be tempered by the available instrumentation available in a laboratory.
Assessment and Management of Risk
Once the ATP has been defined and the analytical technique selected, the potential method can be designed in outline. To ensure that the method is reliable and meets the requirements in the ATP, a risk assessment should be undertaken including the overall procedure. The stimulus paper (1) recommends the use of process mapping the stages of the procedure (as a method is equated to a process) or Ishikawa (fishbone) diagrams to identify potential variables that could have an impact on determining a reliable, reportable result.
A process map should include all stages of an analytical procedure such as sampling, transport of the sample to the laboratory, storage, sample preparation, instrumental analysis, interpretation of results, and calculation of the final reportable value. Variables should be identified against this process map, for example, has a homogeneous sample been taken at the sampling stage or does separation occur during the transport of the sample? A way of ranking these variables, mentioned in the stimulus paper, is a technique known as control, noise, experimental (CNX). Each variable is ranked as a factor to be controlled (C), those that are noise (N), and the ones that need to examined experimentally (X). Naturally, for a regulated industry all of these classifications should be justified and documented. Further risk analysis tools can be used to screen variables to reduce the number of experiments.
Design of Experiments and Robustness Studies
Ideally, this part of the process will be automated using a software application that can control the instrument, acquire and process data, and then refine the experiment. In this phase of the work, variables that could have a large influence on the procedure are identified and examined in detail to determine the design space of the method. Space does not permit me to go into detail on design of experiments (DoE), but there are several papers published on the subject (20–22).
Analytical Control Strategy
The strategy and justification for the approach to control the variables that influence the generation of reliable results and reduce the overall uncertainty of the procedure can be outlined as follows:
- Size of sample to be taken
- Size and number of aliquots for the procedure
- Number of replicate determinations (such as multiple preparations or multiple injections) and how these will be treated to calculate the reportable value
- Impact of instrumental variables on selectivity of the method and how they need to be controlled
- Control of the pH value of mobile-phase buffers for liquid chromatography–mass spectrometry (LC–MS) analysis
- Should LC–MS mobile phases be made up volumetrically or gravimetrically (especially if the percentage organic component of the mobile phase is a key variable)?
- Development of the system suitability test acceptance criteria to determine that a procedure is fit for purpose on the day of analysis
The analytical control strategy can be updated with results from the other two stages of the procedure life cycle.
Knowledge Management
Throughout the method development process, data are generated and are turned into information, which is then transformed into knowledge about how the method operates and what are the key variables. This part of the process enables the understanding of the procedure and is vital to understanding why certain decisions were made about the development.
The key message in the method development and understanding stage of the life cycle is to think before you act: Define what the procedure is required to measure and the matrix in which the determination is to be made. Then spend time looking at the design of the analytical method and conducting experiments to understand and control the key variables. Experience of the analysis of structurally related compounds can be an input here.
Documenting the Analytical Procedure
From this work, the written description of the analytical procedure will be documented and authorized. It needs to have sufficient detail and rationale for the approach to allow an analyst not familiar with the development to set up and run the method. In light of the procedure performance qualification results, there may need to be an update of this document. This stage of the process is the end of the method development work, and now we can move on to method validation.
Validation and Use of the Method
In this section we will consider the validation of the method (procedure performance qualification), its transfer to another laboratory, and classification of the changes that could be made to the procedure and their impact on revalidation.
Procedure Performance Qualification
This is the verification of the performance of the analytical procedure (either a new one or a revised procedure) against the requirements of the ATP. This requires a predefined plan or protocol that outlines the work to be done to demonstrate that the method is fit for its intended use. One aspect of the protocol is how the data will be acquired, interpreted, and compared with predetermined acceptance criteria explicitly stated in the ATP. The key elements from the previous stage's work are the control of key variables, understanding of the way the procedure works (which has been derived from the design of experiments), the robustness of the method, and overall knowledge and understanding of the method.
The work should be performed by a user laboratory under actual conditions of use to comply with existing GMP regulations (23). If the method development stage has been performed well, then this work should merely be a confirmation of that. However, there may be cases where further controls need to be put in place to ensure that reliable reportable results will be obtained, in which case the analytical control strategy will be updated.
When completed, the work will be documented in a report. This report will also outline the work to be done when establishing the procedure in another laboratory, referred to as the local analytical control strategy in the stimulus paper (1).
Procedure Verification
To ensure that an analytical procedure remains in control during the operational use in a laboratory, the main performance indicators should be trended routinely to allow proactive intervention when there is an out-of-expectation (OOE) or out-of-trend (OOT) result. This is done to reduce the number of out-of-specification (OOS) results. Some unexpected results may occur from a variable that was not identified or studied adequately during the method development stage of the life cycle.
Investigation of the problem should identify the root cause of the issue and corrective and preventative action plans should be developed to stop this from occurring again.
Changes to the Analytical Procedure
In addition, there may be time when the analytical procedure can be changed to improve the overall performance of the method. In these cases, the nature of the change indicates what actions should be taken. The stimulus paper defines five types of change:
- Change type 1: Changes within the existing design space of the procedure are considered adjustments and no revalidation is required.
- Change type 2: Changes outside of the proven ranges require confirmation that the method works according to the ATP, but that full redevelopment of the procedure is not required.
- Change type 3: This change occurs if the procedure is to be used in a different environment and is typically treated as a procedure transfer.
- Change type 4: This change requires a new procedure, but the ATP remains the same. In these instances the method development stage is repeated.
- Change type 5: These changes where the ATP is redefined require a full redevelopment and procedure performance qualification.
Now you can also see the importance of the method development stage of the life cycle and knowing the design space. You can reduce the amount of revalidation work when the procedure is used operationally.
Enter Stage Right: FDA Guidance for Industry Number 1
In February 2014, the FDA issued a draft guidance for industry on analytical procedures and methods validation for drugs and biologics (2). This is the update of an existing draft guidance issued in 2000 titled "Analytical Procedures and Methods Validation" (24). It has taken 14 years to update and replace one draft guidance with another one on the same subject. The author wonders if the USP stimulus paper stimulated the FDA action. The FDA guidance aims to complement ICH Q2(R1), which is essentially the same as USP <1225>. As we have seen above, the scope of the ICH document only covers the validation of a procedure and not the journey from concept to realization and ongoing performance verification.
Let's look in some more detail at what is proposed by the FDA. This is not a comprehensive discussion and you are encouraged to read the draft guidance (2) to understand it further.
- There is a section on method development that consists of three paragraphs that paraphrase the method development section in the stimulus paper (1), but the FDA draft guidance lacks the detail of the USP publication. However, the FDA emphasizes the importance of method development and experimental design.
- There is a section on the content of an analytical procedure that is good and would form the basis of a template or checklist for most laboratories. Topics presented include the scope of the method, the instrumentation used, reagents and standards, and calculations and data reporting. There is a statement that you should describe analytical procedures in sufficient detail to allow a competent analyst to reproduce the necessary conditions and obtain results within the proposed acceptance criteria. However, most of this section is in the earlier draft guidance (24).
- The importance of reference standards is emphasized for both traditional small molecules and biologics.
- There is a discussion of statistical analysis and models to evaluate the validation results against predetermined acceptance criteria.
- Finally, there is a section on life-cycle management of analytical procedures. This is perhaps the most perplexing section in the whole document. First, it is placed in the wrong place in the document. If you are going to discuss life cycles, you'll do it at the beginning to set the scene rather that drop it in at the end. Second, the FDA's view of a life cycle is stunted and only begins after the method validation. Surely, a life cycle is from conception to death — perhaps I've been reading the wrong dictionary.
In comparing the USP stimulus paper (1) with the updated FDA draft guidance for industry (2), in part, the FDA document appears as a high-level précis of the USP approach but without the detail. Key parts of the method life cycle are omitted, but there is a good section on what elements should be found in an analytical procedure; however, as noted above, this is more or less a repeat from the earlier draft guidance (24). Yet, the stated aim of the FDA guidance is for validation of analytical methods that will be included in regulatory submissions — if this is an important topic why was the earlier draft guidance not finalized? Why wait until now and issue a document that is so out of date?
One major problem is that the FDA draft guidance references ICH Q2(R1)(6). In my opinion, ICH Q2 is not fit for purpose and needs to be revised to reflect a life-cycle approach similar to the USP stimulus paper (1).
Enter Stage Right: FDA Guidance for Industry Number 2
September 2013 saw the release of the draft FDA guidance for industry on bioanalytical method validation (3); this was a revision of the existing guidance that was issued in 2001 (8). For those that do not know, bioanalysis involves the measurement of therapeutic agents, their metabolites, and sometimes endogenous compounds in biological fluids that typically (but not exclusively) include plasma and urine. The samples for analysis come from animal and human studies for the development of the therapeutic agent. The types of therapeutic agent are either small molecules with molecular weights up to ~2500 Da or biologicals such as peptides, proteins, and antibodies with molecular weights in excess of 5000 Da. Analysis of biologicals typically uses ligand binding assays such as immunoassay or fluorescence-activated cell sorting (FACS), and for small molecules the analytical technique of choice is LC–MS. The FDA guidance document is divided into two sections, one dealing with chromatographic methods and one for ligand binding assays, I will focus on assays for small molecules in this column.
Usually, the amounts to be determined are known from the product specification and have a short dynamic range, such as 50–150% or 75–125% of the nominal amount for the active ingredient, in contrast to impurity analysis that is between 0.05% and 0.5% of the nominal amount of the active component. So in quality control (QC) analysis of a product, batches with many different analytical methods will be used, some will be specific to the product and some will be general analytical techniques applicable to all products — for example, loss on drying.
In contrast, there are relatively fewer bioanalytical methods, but they are applied more intensively to samples from both nonclinical and clinical studies. A clinical study can generate up to 5000 samples for analysis depending on the complexity of the study design and its objectives. Instead of a narrow concentration range as found in a product specification, bioanalytical assays can cover 2–4 orders of magnitude. This is especially true if a drug is given intravenously because the assay will follow the time course of the absorption, distribution, and elimination of the drug in the body, often until the drug is no longer detected.
The major difference of a bioanalytical method is that there is no specification for the sample to be within, but the concentration of a drug is determined over a wide dynamic range. This range is determined by factors such as the amount of drug administered, the route of administration, the dosage form, and how the drug is distributed in and metabolized by the body. The dynamic range of a bioanalytical method can be over two orders of magnitude if the drug is administered intravenously. This brings up the following question: Do bioanalysts dilute samples to get the concentration to a smaller dynamic range where control is better?
Development of Bioanalytical Methods
The focus of the FDA guidance is mainly on validation of a method. The development phase barely gets a mention, two paragraphs in fact on page 5, where the steps and variables in the method need to be investigated to see their impact on the overall method performance. One area that the guidance focuses on in development is that matrix effects from the biological sample are eliminated as much as possible. However, the major flaw with the document is that there is no structure to the planning of and approach to method development, which should make method validation and use easier. Again, similar to the first draft FDA guidance, please read the document to gain a full understanding of the document because I can only summarize key points in this column.


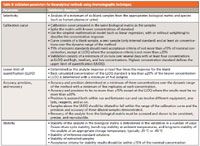
Table II: Validation parameters for bioanalytical methods using chromatographic techniques
Validation Parameters for Bioanalytical Methods
The validation parameters for a bioanalytical method and control parameters when the method is used to quantify analytes in study samples are summarized in Tables II and III, respectively. Some of these parameters are similar to those used in QC laboratories, but some are specific to bioanalysis such as the system suitability, quality control sample acceptance criteria, and the ability to drop calibration curve standards.



Table III: Operational control parameters for bioanalytical methods
Incurred Sample Reanalysis
One major difference between QC analysis and bioanalysis is that with bioanalysis, samples generated for method validation studies are prepared by spiking analytes into a biological matrix. Typically, the solution used to spike the drug concentrations is organic such as methanol or an organic–aqueous solution, depending on the water solubility of the compounds being spiked. This is a potential problem source because the method validation samples and the QC samples used to monitor the performance of the assay when it is applied may not be characteristic of the samples taken from the animals and humans being studied. This problem of the difference between in vivo or incurred samples versus the in vitro or spiked samples has been debated since the first of the AAPS and FDA consensus conferences in 1990. There are many reasons for the differences between spiked and incurred samples, including protein binding, conversion between parent drug, and metabolites and matrix effects.
At the third AAPS and FDA Bioanalytical Method Validation workshop held in 2006 it was indicated that there should be analysis of incurred samples. Incurred sample reanalysis is mainly limited to pharmacokinetic studies to ensure that the conclusions drawn are based on robust bioanalytical data, and it is currently a requirement only in the United States. Early in the study analysis, 20 samples are selected for reanalysis that are close to the highest and lowest concentrations measured from several subjects and 67% of the reanalyzed results should be within 20% of the original results. These results are reported in a separate table from the main study results. The proposal in the draft guidance (3) is that 7% of study samples are reassayed with the same acceptance criteria as above.
Summary
The aim of the USP stimulus paper (1) was to ensure that analytical procedures, through a structured approach to method development and quality by design, become more robust. This approach should result in fewer resources spent troubleshooting problems when the procedure is used for sample analysis or investigating out-of-specification results. Moreover, it should provide greater confidence in the final results generated. This approach should also result in quicker procedure performance qualification (method validation) and procedure transfers, which, in turn, should allow the resources to be reinvested into the method development stage of the life cycle to ensure that procedures are truly robust. In my view, this is an elegant and integrated approach encompassing the whole of the life cycle of an analytical method.
In contrast, both of the FDA draft guidance documents fail on two major issues. First, there is inadequate consideration to designing quality into a procedure via a structured and risk based approach to method development. Second, there is little and poor emphasis on the need for a proper method life cycle (2,3). The failure of the FDA to adequately consider designing quality into analytical methods via a life cycle goes against the principles of ICH Q8 (16) and the failure to incorporate effective risk management in both documents goes against the principles of ICH Q9 (15). Therefore, it raises the following question: Quis custodiet ipsos custodies? (Who guards the guardians?) If the FDA can't or won't include these principles into their guidance documents, who will?
Now, let us turn to both quality control and bioanalytical laboratories implementing the principles of method development outlined in the USP stimulus paper (1). The life-cycle approach requires that laboratories change their current approach, which places more emphasis on the validation of methods rather than development. To do this requires an understanding in experimental design techniques and also the investment in software and instrumentation (if not already available) to design and perform the experiments. However, this will be offset by using more robust methods with a known design space with fewer problem methods and results to investigate. Can laboratories make the jump from the status quo to a better future?
References
(1) G.P. Martin et al., "Lifecycle Management of Analytical Procedures: Method Development, Procedure Performance Qualification, and Procedure Performance Verification," Pharmacopeial Forum39(5) September–October 2013, available at: www.usp.org.
(2) US Food and Drug Administration, Draft Guidance for Industry, Analytical Procedures and Methods Validation for Drugs and Biologics (FDA, Rockville, Maryland, 2014).
(3) US Food and Drug Administration, Draft Guidance for Industry, Bioanalytical Method Validation (FDA, Rockville, Maryland, 2013).
(4) International Conference on Harmonisation, ICH Q2A, Text on Validation of Analytical Procedures (ICH, Geneva, Switzerland, 1994).
(5) International Conference on Harmonisation, ICH Q2A, Guideline on Validation of Analytical Procedures: Methodology (ICH, Geneva, Switzerland, 1996).
(6) International Conference on Harmonisation, ICH Q2(R1), Validation of Analytical Procedures: Text and Methodology (ICH, Geneva, Switzerland, 2005).
(7) V.P. Shah et al., Pharmaceutical Research9, 588–592 (1992).
(8) US Food and Drug Administration, Guidance for Industry, Bioanalytical Method Validation (FDA, Rockville, Maryland, 2001).
(9) General Chapter <1225> "Validation of Analytical Procedures" in United States Pharmacopeia 35–National Formulary 30 (United States Pharmacopeial Convention, Rockville, Maryland, 2011).
(10) General Chapter <1224> "Transfer of Analytical Procedures" in United States Pharmacopeia 35–National Formulary 30 (United States Pharmacopeial Convention, Rockville, Maryland, 2011).
(11) General Chapter <1226> "Verification of Compendial Procedures" in United States Pharmacopeia 35–National Formulary 30 (United States Pharmacopeial Convention, Rockville, Maryland, 2011).
(12) General Notices in United States Pharmacopeia 35–National Formulary 30 (United States Pharmacopeial Convention, Rockville, Maryland, 2011).
(13) International Conference on Harmonisation, ICH Q10 Pharmaceutical Quality Systems (ICH, Geneva, Switzerland, 2008).
(14) European Commission Health and Consumers Directorate-General, EudraLex: The Rules Governing Medicinal Products in the European Union. Volume 4, Good Manufacturing Practice Medicinal Products for Human and Veterinary Use. (Brussels, Belgium, 2013), chapter 1.
(15) European Commission Health and Consumers Directorate-General, EudraLex: The Rules Governing Medicinal Products in the European Union. Volume 4, Good Manufacturing Practice Medicinal Products for Human and Veterinary Use (Brussels, Belgium, 2013), chapter 7.
(16) International Conference on Harmonisation, ICH Q9, Quality Risk Management (ICH, Geneva, Switzerland, 2008).
(17) International Conference on Harmonisation, ICH Q8, Pharmaceutical Development (ICH, Geneva, Switzerland, 2008).
(18) J.M. Juran, Juran on Quality by Design: The New Steps for Planning Quality into Goods and Services (Free Press, Simon and Schuster, New York, 1992).
(19) US Food and Drug Administration, Guidance for Industry, Process Validation (FDA, Rockville, Maryland, 2011).
(20) P. Nethercote and J. Ermer, Pharm. Technol.36(10), 74–79 (2012).
(21) R. Kormany, H.-J. Reiger, and I. Molnar, Recent Developments in HPLC and UHPLC supplement to LCGC Europe, 14–19 (2013).
(22) E. Rozet, P. Lebrun, B. Debrus, B. Boulanger, and P. Hubert, TrAC, Trends Anal. Chem.42, 157–167 (2013).
(23) Current Good Manufacturing Practice for Finished Pharmaceutical Goods, 21 CFR clauses 211.194(a)(2) and 211.194(b) (U.S. Government Printing Office, Washington, DC, 2008).
(24) FDA draft Guidance for Industry, Analytical Procedures and Method Validation (2000).
R.D. McDowall is the Principal of McDowall Consulting and the director of R.D. McDowall Limited, and the editor of the "Questions of Quality" column for LCGC Europe, Spectroscopy's sister magazine. Direct correspondence to: spectroscopyedit@advanstar.com


R.D. McDowall












