Microwave-Induced Combustion for ICP-MS: A Generic Approach to Trace Elemental Analyses of Pharmaceutical Products
A general MIC–ICP-MS method has been successfully developed for the assay of elemental impurities in pharmaceutical products.
Microwave-induced combustion of samples followed by inductively coupled plasma–mass spectrometry measurements (MIC–ICP-MS) is demonstrated to be a reliable and broadly applicable testing method for trace- and ultratrace-level analyses of USP Class 1 and Class 2 defined elements in pharmaceutical products. This method is based on pressurized closed-vessel combustion of samples. The released gases are absorbed in a suitable solution and, if necessary, the remaining oxidized portions of samples are further wet-extracted by reflux that produces complete digestion of samples. The digestion procedure is designed to accommodate for analysis by ICP-MS by using only a limited amount of nitric acid as the absorbing solution, which provides means to avoid large dilution factors and mass interferences. The results demonstrated that the MIC–ICP-MS technique far exceeds the USP detection limit requirements and recovery efficiencies.
Elemental impurities (heavy metals) in pharmaceutical products are introduced by various sources and phases: raw materials, active pharmaceutical ingredients (APIs), formulation agents, solvents, reactors, catalysts, transferring pipelines, and other equipment used during production. Recently, the United States Pharmacopeia (USP) identified and set limits on the concentration of a number of elements in pharmaceuticals and categorized them into Class 1 and 2, as defined by the proposed USP method <232> (1). The Class 1 elements include As, Hg, Cd, and Pb; the so-called "Big Four" that are described as known or strongly suspected human toxicants and thus should be essentially absent (<1.5 μg/g). The Class 2 elements, which include Cr, Cu, Mn, Mo, Ni, Pd, Pt, V, Os, Rh, Ru, and Ir, are relatively less toxic yet commonly found impurities. Such impurities also could affect the stability and shelf life of drugs because of their catalytic activities.
Elemental impurities continue to be assessed by the concomitant visual comparison test (2). This method requires a sample preparation step that involves ashing at 600–800 °C followed by acid digestion of the residues. The considerable evaporation of some elements (As, Hg, and so forth) during ashing (3) and the inability to form colored complexes with sulfide ions by some elements (As, Cd, Hg, and so forth) have prompted the development of improved sample preparation procedures in combination with instrument-based analytical methods such as inductively coupled plasma–optical emission spectroscopy (ICP-OES) and inductively coupled plasma–mass spectrometry (ICP-MS), which demonstrate improved sensitivity, elemental specificity for most of the elements in the periodic table, and a wide linear dynamic concentration range.
Significant progress and advancement (4–7) have been achieved in this field. USP method <233> describes a sample preparation decision tree for ICP-OES and ICP-MS assays, including dissolution in aqueous medium, dissolution in organic solvent, and closed-vessel microwave pressure digestion (8). USP is pending to issue a final official recommendation on alternative methodologies for elemental impurities analysis. Although such efforts have greatly improved the accuracy, sensitivity, and elemental specificity, a number of considerations still remain, ranging from the general applicability of the proposed methods to the wide range of organic compounds used in the pharmaceutical industry, the technical specifics such as the availability of matrix matching certified standards (matrices consisting of organic solvents), analyte loss because of metal ion complexation by organic ligands, high residual carbon contents because of incomplete digestion, and high blank values.
Because it has been used for more than 100 years, the pharmacopeia ashing–acid digestion methodology merits a revisit by virtue of its ability to liberate elemental impurities from nearly all types of organic pharmaceuticals into an acidic aqueous solution suitable for atomic spectroscopic and chromatographic assay. The microwave-induced combustion (MIC) procedure advances the conventional ashing–combustion technique by integrating the combustion and acid digestion into a single closed-vessel system. This technique uses high-power microwave radiation to trigger the combustion of samples in an oxygen-pressurized thick-wall quartz vessel (Figure 1). In the subsequent refluxing cycle, a medium-to-low power microwave radiation is applied to create high-pressure, high-temperature conditions for further digesting residues and recovering analytes in the presence of an absorbing solution. The MIC procedure has proven to be effective for digesting hydrocarbon polymers (9) and nuclear-grade graphite materials (10), for which the current digestion techniques can be difficult and complex.


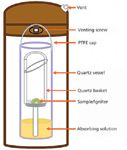
Figure 1: MIC digestion apparatus.
Modern drug products use increasingly sophisticated, carefully designed protective exterior layers and scaffolds for drug delivery. For example, many oral medicines have enteric coatings that prevent release of medication before they reach the small intestine so as not to unsettle the stomach. These enteric coatings usually consist of synthetic polymers or biopolymers such as poly(vinyl acetate phthalate), methyl methacrylate–methacrylic acid copolymers, fatty acids, waxes, shellac, and polysaccharides. The aliphatic skeletons of these polymers are often hydrolytic and oxidative resistant and tend to carbonize at elevated temperatures in the absence of an oxidative atmosphere, leading to high residual carbon content (11). Initial efforts by our laboratory to digest coated aspirin tablets using microwave pressure digestion in concentrated HNO3 or HNO3–H2SO4 mixtures at 220 °C and 80 bar have been hampered by the chemical inertness of these coatings.
The prospect that MIC could mineralize diverse organic matrices including APIs and excipients into a simple aqueous matrix that is suitable for ICP-MS assay prompted us to launch a feasibility study for surveying critical elemental impurities of pharmaceutical products, in particular those with delivery excipients. Four over-the-counter aspirins were selected for evaluation. The solid formation of pharmaceutical entities can be assessed readily by thermogravimetric analyses (12). The thermal analysis data revealed that these drug entities all exhibit multistage degradation, revealing that the enteric coated aspirins are, by themselves, a very complex system comprising an organic polymer coating, a small organic molecule API, buffering ingredients, and other formulation excipients. Despite having a common API (that is, acetylsalicylic acid), each aspirin brand exhibited different mass loss patterns, indicative of a unique formulation. In this work, the efficacy of the MIC–ICP-MS method for elemental impurity assay was investigated, and the results demonstrated that this method is superior to the existing methods in terms of broad applicability, accuracy, sensitivity, and elemental specificity.
Experimental
A single aspirin tablet, approximately 400–700 mg, was crushed into small pieces using a mortar and pestle. The crushed sample was then enveloped with an ashless filter paper and placed onto the quartz basket, to which a 1.5 M NH4NO3 solution (an igniter) was added drop-wise to soak the sample before it was loaded into the rotor 8SXQ80 vessel of the Multiwave 3000 microwave unit (Anton Paar, Österreich, Austria) and pressurized to 20 bar of high-purity oxygen. The absorbing solution in the quartz vessel that was used was 5 mL of 20% (v/v) HNO3. This procedure was designed specifically so that the final acid matrix in the sample was 2% (v/v) nitric acid in a final volume of 50 mL and no further dilution was required for analysis. The microwave digestion program was designed to ensure sufficient cooling after the combustion and refluxing stages. Sample digestions were prepared in triplicates using an entire tablet for each replicate. Every MIC digestion was carried out with a digestion blank, as well as a calibration verification standard that was used to check for instrument drift.
A PerkinElmer (Waltham, Massachusetts) ELAN DRC II ICP-MS system equipped with a Cetac ASX-520 autosampler was used to measure the concentrations of the selected elements in the sample solutions. The instrument analytical conditions and parameters are listed in Table I. The isotopes that were used in the measurements were 51V, 52Cr, 55Mn, 60Ni, 65Cu, 75As, 98Mo, 101Ru, 103Rh, 105Pd, 114Cd, 189Os, 191Ir, 195Pt, 201Hg, and 208Pb, but multiple isotopes were monitored for non-monoisotopic elements to check for potential mass interferences. The internal standards that were used were 45Sc, 89Y, 115In, and 159Tb. Spike-recovery tests were performed at the digestion step on the sample tablets and the sample solutions. The former would test for any loss of elements during digestions, which is critical for evaluation and validation of the digestion method against the conventional USP procedures.


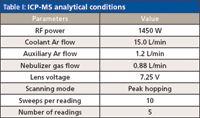
Table I: ICP-MS analytical conditions
Results and Discussion
To minimize sampling bias, the entire enteric coated pill was taken into account. These tablets weighed approximately 400–700 mg each, which is a substantial amount for a typical MIC operation. A small amount of NH4NO3 was used as an igniter, which is enough to sufficiently generate high temperature–high pressure conditions. Figure 2 shows a typical MIC real-time run condition profile. An initial high-power microwave irradiation (1400 W) ignites the sample, leading to a rapid pressure increase that actually causes momentary power cut-off. In the reflux cycle, the internal pressure built up to 80 bar while the temperature reached 220 °C. This is a rather favorable condition for digesting the combustion residuals and for refluxing recovery of analytes from internal walls and surfaces. The digestions were allowed to cool down to room temperature before venting off the residual gases. An entire MIC run takes approximately 2 h and can digest up to eight samples at a time using the 8SXQ80 rotor. The resulting digestion solutions were all visually clear with residual carbon content of <1%. These results demonstrate that the MIC conditions employed here are effective in completely digesting these enteric-coated drugs, regardless of their formulations or chemical structures. The digestion protocol can be optimized further for shorter digestion times (higher throughput) by reducing the sample size, altering the absorbing solution compositions, and increasing the igniter amount.



Figure 2: Real-time power, temperature, and pressure profiles of the MIC digestion conditions.
The overall elemental accuracy and specificity of the MIC–ICP-MS method were evaluated in two steps: pre-MIC digestion spike and post-digestion spike. The latter was carried out by spiking the digestion solutions at concentrations of 10 and 100 μg/L. These levels were chosen to represent the concentration in the solid tablets at 1 and 10 μg/g. The major matrix element in the aspirin samples is carbon, which ranges from 31 wt% of Aspirin C to 49 wt% of Aspirin D (Table II). Table III shows the spike recovery data for both Aspirin A and B digestion solutions. The spiked Aspirin A solution demonstrated close-to-complete recoveries (95–103%) for all elements of interest, at both low and high spike levels. Similar recoveries were achieved for the Aspirin B solution. These data suggest that the space charge effect in the mass spectrometer, mainly caused by the matrix element concentration, is minimal. The low matrix effect could be accredited to the fact that combustion gasifies organic matrices into CO2 and CO. Such gas molecules have limited solubility in the acidic absorbing solution and subsequently are released to the atmosphere.


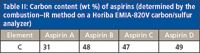
Table II: Carbon content (wt %) of aspirins (determined by the combustionâIR method on a Horiba EMIA-820V carbon/sulfur analyzer)
To evaluate the spike recovery of the MIC procedure, the absorbing solution was spiked at levels equivalent to 1 and 10 μg/g for each element in the solid samples before initiating the procedure. Table III shows the MIC spike recovery results for Aspirin A. All Class 1 elements (As, Cd, Hg, and Pb) were recovered at 96–102% after undergoing the MIC digestion process for both spike levels, and most of the Class 2 elements (V, Cr, Mn, Ni, Cu, Mo, Ru, Rh, Ir, Pd, and Pt) were recovered at 86–112%. The spike recovery for osmium, however, was unusually high at over 200%. This is suspected to be because of the formation of osmium tetroxide (OsO4) in the nitric acid medium during the digestion process, which could cause false high readings because of the increased amount of gaseous tetroxide reaching the plasma (13). Therefore, it is recommended that a longer nebulization time is used to reach a steady state before data acquisition or hydrochloric acids be used for osmium analysis. Overall, the spike recovery results presented here meet the USP proposed acceptance criteria of 80–150%. The efficient spike recovery demonstrated by the MIC method is accredited to the use of a closed-vessel system along with a combustion process that decomposes organic matrices and their functional groups, such as carboxylic (–COO-), thiol (–SH), thioether (RSR), and cyano (–CN-) groups, which are known to have strong binding abilities toward Hg, Pt, Pd, and Sn that can decrease the recovery of these elements.


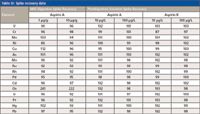
Table III: Spike recovery data
It is worth noting that for the precious group metals (Ru, Rh, Pd, and Pt), an equal or slightly lower recovery was found in the high-dose spiking experiment, as shown in Table III. It is suspected that the solubility of these elements was reduced because of the high dilution factors (100×) of the Cl- ion, which originally was added in the standard solutions as stabilizers. In general, the HCl system can show better recovery for these elements, including osmium, in comparison to the HNO3 system. Although the addition of the Cl- ions to the current absorbing solution may improve the recovery of these elements, the chlorine isotopes 35Cl and 37Cl are known to form polyatomic species such as 40Ar35Cl+, 38Ar37Cl+, 35Cl16O+, and 37Cl16O+ species, which can complicate the measurement of isotopes such as 75As+, 51V+, and 53Cr+. However, with the commonly available collision reaction interface and dynamic reaction cells, in many ICP-MS instruments, such mass interferences can be readily resolved. The other alternative for ICP-MS analysis is to separate the measurement of the precious groups elements from the rest of the elements.
Table IV summarizes the results to the ICP-MS analysis of the four samples. The detection limits (DL), which were calculated from 3σ of the digestion blank (n = 10), are also shown in the table. The DLs were found to range from 0.002 μg/g to 0.006 μg/g for all the elements of interest. It should be emphasized that the absolute DL may not be necessary for such a general specification purpose. It is more useful and effective to perform a single quantification limit test. The low DL achieved here likely is attributed to the low blank values of the digestion media because of the use of dilute HNO3 and the small dilution factor that was involved in the analysis. To a lesser extent, this dilution factor is attributed to the use of quartz vessels, which have smoother surfaces (14), compared to PTFE vessels, which have rough and porous surfaces that can result in high carryover, or to the metal alloy vessels that are used in the conventional oxygen bomb technique (15). Neither individually nor collectively do the elemental impurities of the investigated aspirin lots exceed the limit set by the USP.


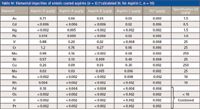
Table IV: Elemental impurities of enteric coated aspirins (n = 3) (*calculated DL for Aspirin C, n = 10)
In general, the current MIC–ICP-MS approach implements a number of features to achieve broad applicability, excellent recoveries, high sensitivity, and specificity for trace elemental impurity assay of pharmaceutical products. These include a combustion process that can readily liberate elemental impurities from organic matrices and avoid the effect of matrix in the analysis; a closed quartz vessel system that minimizes the analyte loss and the surface carryover that often are encountered by the use of PTFE vessels as a result of the sintering-caused surface roughening; a high-pressure, high-temperature refluxing cycle ensuring thorough washing-off of analytes from the internal surfaces by the constantly refluxed absorbing solutions; a simple H2O–HNO3–O2 digestion system that does not require the use of H2SO4, HCl, or organic solvents that may contribute to the mass interference or high background levels; a digestion medium containing no metal ion-complexing ligands but simple inorganic counter ions such as NO3-, Cl-, and SO42-; readily available NIST traceable certified standard solutions for calibration without the need for preparing sophisticated matrix matching standards; and the ability to design the digestion and absorbing solutions to accommodate for the analysis of interest.
Conclusion
A general MIC–ICP-MS method has been successfully developed for the assay of elemental impurities in pharmaceutical products, as demonstrated using over-the-counter enteric-coated aspirins of various brands. Recoveries of 100 ± 5 % and detection limits below 0.01 μg/g have been achieved for most of the Class 1 and Class 2 elemental impurities, which exceeded the USP requirements (recovery 80–150% and μg/g-level limit) (1).
The pharmaceutical industry has been actively exploring and deploying multifarious drug delivery systems for a variety of purposes: controlled-release, site-direction, protection, and stabilization of APIs. These delivery systems frequently rely on various biocompatible and biodegradable excipients to achieve the desired functions, such as synthetic and biopolymer hydrogels (polyols, polyesters, polyethers, and polycarboxylic acids of linear, hyper-branched, and dendritic architectures), and solid lipid nanoparticles. There are also a widespread use of supplementary dietary products and the increasing popularity of herbal medicines, all of which require specifying elemental impurity levels (USP <2232>). The chemistry perspective of these organic matrices is nothing more than C-H frameworks decorated with heteroatoms such as O, N, S, and P, which are conceivable, although expensive, oxygenated or nitrogenated (or both) fuels. In principle, MIC in combination with ICP-MS (or ICP-OES) may well be used to meet these requirements. These aspects currently are under investigation and their results will be published elsewhere.
It also should be pointed out that the high recovery and high sensitivity quantification of trace elements offered by MIC–ICP-MS is of great advantage for the specification of increasingly costly APIs, which are often produced in small quantities, and for screening and designing novel therapeutic and diagnostic agents in the field of metal-complex medicines ("elemental medicines"), which is an emerging area of chemistry aimed at the treatment and understanding of diseases that are currently intractable.
Acknowledgments
We thank analysts Kevin O'Brien, Susan Dunn, and Oanh Pham for their assistance in this research.
References
(1) Elemental Impurities–Limits, Pharm. Forum 36(1), Chapter <232> (2010).
(2) D, Ciciarelli, D. Jakel, and E. Konig, Pharm. Forum 21(6), 1638–1640 (1995).
(3) S. Lira, P. Brush, L. Senak, C. Wu, and E. Malawer, Pharm. Forum 34(6), 1613 (2008).
(4) N. Lewen, S. Mathew, M. Schenkenberger, and T. Ragllione, J. Pharm. Biomed. Anal. 35, 739–752 (2004).
(5) S. Lira, P. Brush, L. Senak, C. Wu, and E. Malawer, Pharm. Forum 34(6), 1613–1618 (2008).
(6) N. Rao and K. Talluri, J. Pharm. Biomed. Anal. 43, 1–13 (2007).
(7) J. Huang, X. Hu, J. Zhang, K. Li, Y. Yan, and X. Xu, J. Pharm. Biomed. Anal. 40, 227–234 (2006).
(8) Elemental Impurities–Procedures, Pharm. Forum 36(1), Chapter <233> (2010).
(9) E. Moraes, M. Mesko, P. Mello, J. Paniz, V. Dressler, G. Knapp, and E. Flores, Spectrochim. Acta B 62(9), 1065–1071 (2007).
(10) X. Wang, G. Bhagat, K. O'Brien, and K. Putyera, 2009 MRS Fall Meeting Symposium V Proceedings, Boston (2009).
(11) G. Arujo, M. Gonzalez, A. Ferreira, A. Nogueira, J. Nobrega, Spectrochim. Acta B 57, 2121–2132 (2002).
(12) N. Redman-Furey, M. Dicks, J. Godlweski, D. Vaughn, and W. Collins, J. ASTM Intl. 2(1), 23–32 (2005).
(13) D.C. Gregoire, Anal. Chem. 62, 141–146 (1990).
(14) H.M. Ortner, H.H. Xu, J. Dahmen, K. Englert, H. Opfermann, and W. Görtz, Fresenius J. Anal. Chem. 355, 657–664 (1996).
(15) G.B. Souza, E.N.V.M. Carrilho, C.V. Oliveira, A.R.A. Nogueira, and J.A. Nobrega, Spectrochim. Acta B 57, 2195–2201 (2002).
Kwan H. Nam, Robert Isensee, Gabe Infantino, Karol Putyera, and Xinwei Wang are with Evans Analytical Group, Syracuse, New York.














High-Speed Laser MS for Precise, Prep-Free Environmental Particle Tracking
April 21st 2025Scientists at Oak Ridge National Laboratory have demonstrated that a fast, laser-based mass spectrometry method—LA-ICP-TOF-MS—can accurately detect and identify airborne environmental particles, including toxic metal particles like ruthenium, without the need for complex sample preparation. The work offers a breakthrough in rapid, high-resolution analysis of environmental pollutants.




Trending on Spectroscopy: The Top Content of 2024
December 30th 2024In 2024, we launched multiple content series, covered major conferences, presented two awards, and continued our monthly Analytically Speaking episodes. Below, you'll find a selection of the most popular content from Spectroscopy over the past year.


