Analytical Efforts Toward Monitoring Groundwater in Regions of Unconventional Oil and Gas Exploration
Special Issues
A mix of analytical methods is required to understand the impact, if any, that UOG activity is having on groundwater.
Gas chromatography (GC), inductively coupled plasma–mass spectrometry (ICP-MS), ICPâoptical emission spectrometry (OES), and other bulk analysis methods are applied to groundwater in proximity to unconventional oil and natural gas extraction activities.
The United States has experienced a dramatic shift in economic influence over the past 10 years with the widespread engineering advances that have allowed unconventional oil and gas (UOG) extraction to become more efficient and cost-effective. Small rural towns have become industry hubs overnight as a result of the hydrocarbons trapped beneath the ground. Educators have increased the number of engineering and technical programs available to students in an effort to meet the demand for a qualified workforce. Stories of multigeneration ranchers becoming millionaires overnight through the leasing of their land and mineral rights puts the television show The Beverly Hillbillies in a more current light.
Regulations related to UOG activity are currently left to each state, which creates disparities in environmental testing and monitoring across the United States. For example, Colorado and Illinois require baseline groundwater testing before drilling commences, while Pennsylvania only suggests baseline measurements in the event that a dispute arises after drilling. The list of organic compounds Colorado has chosen to monitor through chromatographic methods are total petroleum hydrocarbons, benzene, toluene, ethyl benzene, and xylenes (BTEX), polycyclic aromatic hydrocarbons (PAH) plus benzo[a]pyrene, and dissolved gases. These analytes have been the focus of analyses for years. Their presence in water is hypothesized to indicate an adverse environmental interaction with hydrocarbon extraction operations. However, their sources can still be convoluted and may not be wholly specific to UOG.
Within the past decade, a mix of analytical methods has been developed or applied to establish an understanding of the impact, if any, that UOG activity is having on groundwater in the vicinity. This article discusses chromatographic methods applied for particular organic compounds and considerations to assist in method development. A section is also dedicated to spectroscopic methods for detection and quantification of metals and ions in water samples that are relevant to UOG activity. Collections from our groundwater research are highlighted in each section to demonstrate the application.
Chromatography for the UOG Field
The ideal situation for creating a method would be to work with a system of known knowns (1) or compounds that are expected to be present and are positively identified. Current researchers must also have the capabilities to work with unknown knowns or the ability to determine unexpected identifiable compounds, and unknown unknowns, compounds unexpected and without standards, Chemical Abstracts Service (CAS) numbers, or absent in databases. For example, these may include proprietary polymers or surfactants developed primarily for the UOG field. A hesitation that may be encountered are known unknowns, which are expected compounds not detected. The internal debates are made up of how confident the compound is to be a “known;” is the concentration too low to detect in the given sample, or is the method inadequate? In the discussion to follow, there are very few “knowns” to be expected when monitoring groundwater possibly impacted by UOG activity.
Gas Chromatography
Gas chromatography (GC) methods have been at the forefront for analysis of organic compounds in groundwater and UOG wastewater (UOGWW) (2). While there is the potential for nonvolatile organic additives such as surfactants to be present, the majority of hydraulic fracturing additives or shale formation compounds of health or environmental concern are GC amenable. In a 2011 Congressional report, 24 organic hydraulic fracturing additives are listed as “Chemical Components of Concern,” of which 23 are GC amenable without the need for derivatization. Some of these include BTEX, diesel, and naphthalene, which have been suggested for baseline measurements by various states.
Numerous Environmental Protection Agency (EPA) and state regulatory methods have been established using GC for these and a multitude of other compounds of concern over the past 50 years. While a mix of regulatory methods can be found that include a subset of these compounds, the lack of a single dedicated standard approach to effectively extract and separate a probable list of compounds in groundwater or UOGWW is a complicating factor that has slowed research. Most officially standardized versions of these methods are less capable of the throughput needed to prepare and analyze a large number of samples in a limited timeframe.
Dissolved Gas Analysis
The earliest efforts to assess the impact of UOG activity on groundwater was through the measurement of dissolved gases, specifically methane, ethane, propane, butane, and pentane (C1, C2, C3, C4, and C5, respectively) in groundwater from regions within close proximity of UOG drilling sites (3). Methane is the most abundant component of natural gas extracted for energy purposes, with ethane and propane comprising the majority of the remaining small fraction. The hypothesis is that if there is a failure in the integrity of the protective casing of the UOG well (4,5) or if induced fractures in the shale create interconnectivity with the overlying aquifer, the natural gas would be the most abundant and mobile species to detect in groundwater.
Two types of methane can be measured in groundwater (6). The most common type found in shallow groundwater is biogenic methane, a by-product of bacterial metabolism. Thermogenic methane is the other type, the primary target of UOG recovery. This methane gas is formed by the presence of decomposing organic matter under high temperatures and pressures over a long period of time (that is, from deep geological formations). Because of the different implications for each type of natural gas, methane measured in shallow groundwater must include further investigations to distinguish between biogenic or thermogenic origins.
The origin of the measured methane can be determined either through isotopic abundances of carbon-13 (13C), deuterium (2H), or the methane to ethane and propane ratio (7). A ratio of methane to higher chain hydrocarbons of less than approximately 100 suggests thermogenic gas (3). Both of these approaches have even been found to not only identify thermogenic methane, but also distinguish between different natural gases produced in different geological formations (3,8,9).
GC separations coupled with flame ionization detection (FID) are most commonly used for the analysis of these light hydrocarbons. Groups measuring these light hydrocarbons do so in a targeted manner, meaning they are tailored specifically for C1–C5 gases and little else. These methods are quite sensitive and selective, but ultimately lack the ability to detect a wide suite of unknown compounds. Sample introduction is performed through either purge-and-trap techniques (10), where the water is purged with an inert gas and volatiles are trapped on a selective sorbent, or using headspace analysis (11), where the sample is heated and agitated to liberate the gas to an open headspace in the vial, which is then sampled. Column selection for this analysis leans toward the use of porous layered open tubular (PLOT) columns, primarily those with a divinylbenzene phase (10,12). As alluded to earlier, PLOT phases possess a great affinity for C1-C5 hydrocarbons, but that affinity is further extrapolated to the C6-C8 linear and branched alkanes and aromatics, which are also present in natural gas, making analysis of these larger hydrocarbons inefficient because of long retention and excessive band broadening. An example of this affinity is demonstrated in Figure 1 with a natural gas standard separated on a PLOT divinylbenzene column and a 5% diphenyl capillary column.


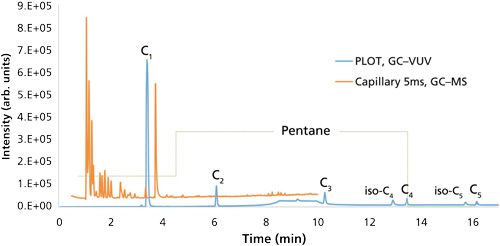
Intricate sample collection further complicates this specific analysis, which typically requires additional measurements, such as isotopic analysis, for results that can enable sourcing of the natural gas in the water (that is, a comparison of the natural gas isotope signature in the water with that from the targeted shale or other sources). Groundwater samples are typically collected from volunteers’ water wells, in which the withdraw rate and consistency will be variable across the population. Agitation, along with the pressure differential on the water once it reaches the surface, can cause the water to degas and skew dissolved gas measurements to below their actual values. For better control during sampling, it is recommended to use a nongas permeable tube with a valve connected to the water well head that flows at a constant rate into an evacuated bladder (for example, an IsoFlask sampling bladder [Isotech Laboratories]). This sampling bladder should be preloaded with a chosen biocide to reduced degradation of the gas by bacteria, on top of being stored at 4 °C for a 14-day maximum holding time before analysis (13).
As a complementary approach, our group has also demonstrated the capabilities of a new spectroscopic detector, the VGA-100 vacuum ultraviolet (VUV) detector (14) (VUV Analytics), which measures gas phase absorption in the VUV and ultraviolet wavelength regions (120–240 nm) to monitor dissolved gases in water. This universal detector offers qualitative gas-phase VUV spectra to accompany the quantitative capabilities of the C1–C5 hydrocarbons, along with N2, O2, and CO2 if interested (15). While this work separated C1–C5 hydrocarbons of three water samples from the Barnett Shale with the HP-PLOT Q column (30 m × 0.32 mm, 20-µm df), the deconvolution capabilities of the acquired spectra could allow the compounds to be quantified in the void volume of capillary columns. More work is needed to interface this detector with the various sampling protocols that exist for measuring natural gas in water to demonstrate its unique qualitative and quantitative capabilities for routine analysis. The blue chromatogram of Figure 1 is the previously discussed GC separation of a natural gas standard with the PLOT divinylbenzene column and VUV detection.
Organic Compounds
In the vast majority of UOG reservoirs, hydraulic fracturing is used to stimulate the formation. The fluids used to open fissures in low permeability shale formations include water, sand, and a small percentage of chemicals. These chemicals are a mixture of acids, bases, salts, organic compounds, and inorganic compounds, which serve myriad purposes. Even though these chemicals make up a small percentage of the liquid used for hydraulic fracturing, it can account for a median of over 10,000 kg (16) in the national average of 2.4 million gallons of water used per UOG well (17). This massive amount of chemicals is trucked to the pad site, stored and mixed onsite, and injected for hydraulic fracturing operations. Then, up to 30% of the water resurfaces during the flowback period before production begins. The storage, use, and collection of these chemicals, mixed hydraulic fracturing fluids, other chemicals involved in equipment cleaning and drilling processes, and the resulting flowback are all possible sources for groundwater contamination through controllable surface activities (18). Casing and cement failures (19) are a subsurface possibility for fluid introduction to the aquifer system, an event with little operator control, but which occurs at varying rates reported to be from 3% (20) up to as many as 12% of wells within the first five years (21).
The majority of hydraulic fracturing additives can be found in lists that have been becoming more populated over the recent years. One of the earliest lists (22) was found in a report by the US House of Representatives Committee on Energy and Commerce. This included over 750 unique additives found in more than 2500 products available to be used for hydraulic fracturing from 2005 to 2009. Additional pertinent information included in the report are the number of products in which the compound is found, a table of additives that are health or environmental risks, and highlights of statistics for use of specific compounds of concern like 2-butoxyethanol. FracFocus (www.fracfocus.org), instated in April 2011, is the national hydraulic fracturing chemical registry. In the US, 28 states require chemical disclosure of hydraulic fracturing fluids, of which 22 are using FracFocus. It currently contains more than 80,000 disclosure documents from more than 1000 companies. This registry provides the operator, location, depths, chemicals, and mixed concentrations used in hydraulic fracturing activities (23). The companies that disclose this information are able to protect trade secrets with the ability to report some additives as “proprietary polymers” or under similar designations. A review of the FracFocus database from January 2011 through February 2013 (16) revealed more than 37,000 logs, which included chemical disclosure of 692 unique ingredients, of which 11% were deemed trade secrets.
GC coupled to mass spectrometry (MS) has been the workhorse used to separate, detect, and possibly identify volatile and semivolatile compounds present in groundwater, after appropriate sample preparation (2). The MS detector is practically a requirement when surveying groundwater for contaminants related to UOG, even when performing targeted analysis for specific compounds. The qualitative information gained from the MS detector is invaluable in confirmation and unknown identification. The potentially complex mixture of compounds in groundwater impacted by UOG has generated false positives when using the suggested FID in EPA methods used for general groundwater (24).
The overwhelming majority of the capability of unknown identification with GC–MS begins with the electron ionization (EI) source (1). The EI source generates diagnostic fragment ions of the compound in a systematic manner. The resulting spectra can then be matched across a number of mass spectral libraries, generated by the National Institute of Standards and Technology (NIST), National Institute of Health (NIH), and the EPA, among others. Another ionization source, chemical ionization (CI), can be used to complement the EI-resulting data. The molecular ion for the compound is generally preserved by CI. CI can be described as a softer ionization technique; therefore it generates fewer fragments and is not used as the primary source for unknown identification. An MS detector capable of CI can also possess the ability for negative chemical ionization (NCI), a selective ionization technique effective toward ionizing halogenated compounds. This selective ionization is a detriment to broad surveying of unknown compounds, but it is a valuable tool for researchers investigating halogenated species.
Fragmentation information from tandem MS (MS-MS) for further confidence in identification can be generated by ion-trap or triple-quadrupole MS detectors. High resolution and accurate mass (HR-AM) analysis of ions for an additional identification vector can be achieved with time-of-flight (TOF) and orbital trap MS detectors. Hybrid MS detectors can also be found to combine the MS-MS capabilities with HR-AM on the back-end, as with a Q-TOF or Q-orbital trap.
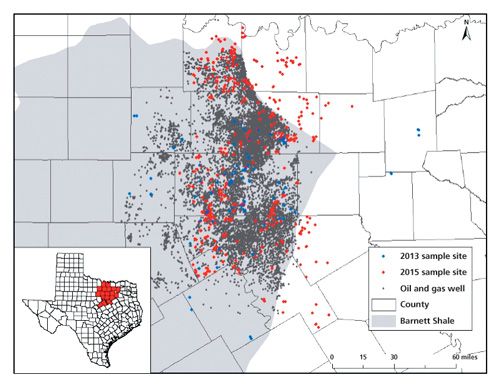
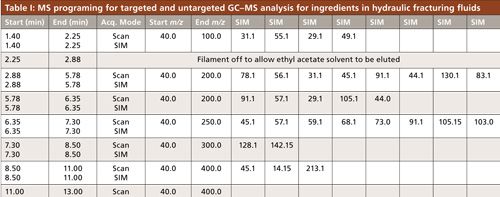
A portion of a Texas well water study conducted by our group included identification of the volatile and semivolatile compounds in groundwater across the Barnett Shale region, shown with sampling locations in Figure 2. A GC–MS method was developed to provide appropriate sensitivity and good sample throughput. The aim of the method was to extract and separate the greatest number of compounds from a small volume of water with minimal sample preparation. Sample preparation on the front end included a 2-mL ethyl acetate extraction from 5 mL of groundwater, shaken 1 min in a screw-top vial. GC–MS analysis was performed using a 30 m × 0.25 mm, 0.25-µm df Rxi-5ms (Restek) column with a single-quadrupole MS detector using an EI source. The “5” column, or 5% diphenyl, 95% PDMS stationary phase, is typically regarded as a general purpose column and has retention characteristics quite comparable to columns in which Kovat’s retention indices were calculated. The retention index of an unknown peak can be a valuable piece of data to narrow down possibilities of detected unknown compounds (25). Separation and MS parameters were set for the detection of 35 target compounds in our method. These compounds were chosen based on their popularity of use, possible health and environmental effects, detection in previous research, and GC amenability. These compounds consisted of various alcohols, aromatics, and other hydrocarbons. MS settings, summarized in Table I, included groups of selected ion monitoring (SIM) events for the base peaks of our target compounds, coupled with full spectral scanning for the confirmation of measured peaks by SIM, as well as the possible identification of unknowns through spectral matching. These acquisition groups were typically around 2 min each in an effort to keep the SIM ion count low to maintain an effective MS duty cycle. The scanning parameters also changed with each acquisition group, in that the concluding m/z increased from 100 to 400 over the time of the separation. The assumption used was that compounds eluted earlier from the column would be lighter than those eluted later; reducing the acquisition window reduces noise in the spectrum.




Initial application of this method detected methanol and ethanol in some groundwater samples. These detections were unable to be quantified with data at the time because of poor retention on the Rxi-5ms column and a fair amount of background noise from permanent gases like N2 and O2 when monitoring their base peak, 31 m/z. This led our team to develop a method to address both of these problems.
A mid-polarity GC column was chosen for the complementary analysis. The team still wanted to maintain the ability to adequately retain linear hydrocarbons if present, so working with a 100% PEG column was nearly out of the question, even though it maintains a great selectivity for these alcohols. The 30 m × 0.32 mm, 1.20-µm df Phenomenex ZB-BAC2 column, developed and marketed for blood alcohol analysis, was chosen for its retention and selectivity for these two alcohols and other solvents, along with the possibility of using a second paired column, the Phenomenex ZB-BAC1 column, for confirmation if needed. The method also incorporated FID to help reduce the background noise while detecting the light alcohols. A static headspace injection technique was chosen to effectively extract the analytes of interest and reduce background. A salt solution was added to the water sample to reduce the solubility of the alcohols in groundwater. Samples were agitated, heated, and injected automatically using an AOC-5000plus autosampler (Shimadzu Scientific Instruments).
In our initial study of Barnett Shale groundwater in 2011 (26), 29 of the 100 samples contained methanol at concentrations as high as 329 mg/L and 12 samples contained ethanol at levels as high as 11 mg/L. These detections had no correlation with distance to UOG wells. Numerous industrial processes use these alcohols, and they can be produced through a range of biological pathways, so identifying the sources for the occurrences was not practical with the limited data.
A follow-up study in 2014 expanded the research to 550 groundwater samples across 13 counties in north Texas (27), shown in Figure 2. Additional compounds were detected in this study in addition to the methanol and ethanol from the previous studies. Alcohols included methanol (35 wells), ethanol (240), isopropyl alcohol (8), and propargyl alcohol (155). Ethanol and propargyl alcohol had a positive correlation with each other. These are both ingredients in hydraulic fracturing fluids and were detected at a higher frequency than expected in the most productive counties based on chi-squared analysis. Chloroform, dichloromethane, and trichloroethylene were detected in 330, 122, and 14 wells, respectively. These chlorinated compounds are not disclosed ingredients in hydraulic fracturing fluid, but have been identified in UOGWW (28) and dichloromethane has been suggested (29) to be present during drilling operations as a degreaser for equipment. The study also found that 381 samples contained at least one aromatic of the BTEX class, with 10 samples containing all four species. Benzene was detected in 34 wells, toluene in 240, ethyl benzene in 22, and at least one xylene isomer in 240 water well samples. The BTEX compounds collectively can be found in hydrocarbon fuels, refined or unrefined, and some are used individually as industrial solvents, even as hydraulic fracturing additives.
All of the compounds mentioned above can be linked directly or indirectly to UOG operations. The fact that these compounds are fairly common in the industrial or agricultural setting in which this research was conducted, renders it impossible to implicate UOG as the source of the contaminants with absolute confidence. It is expected that the only definitive manner in which to conclusively attribute UOG as a source of groundwater contamination would be through the detection of proprietary tracers (30), suggested to be fluorinated compounds exotic enough to assist each company with MS detection for internal monitoring.
Spectroscopy
Chromatography has been at the forefront of advanced analytical chemistry to tackle the challenges of analyzing complex mixtures related to UOG that possibly could be encountered during research. The previously discussed approaches are appropriate when identifying individual compounds, but there are situations when monitoring of bulk chemical classes yields adequate information. Many metals and ions can also be determined spectroscopically (31). For the most part, the operational costs for these methods are less than chromatography–MS methods, less technical to operate, and can even be performed portably. Spectroscopic approaches associated with UOG have included UV–vis spectroscopy, infrared (IR) spectroscopy, and optical emission spectroscopy (OES). Yet, many of these methods can fall victim to interferences from chemically similar compounds or ions since they are being measured in bulk solution without prior sample preparation. These methods are also typically intended for oil field waste waters or produced water, both of which commonly contain higher concentrations of the analyte than ever expected in compromised groundwater.
Absorbance methods measured in the UV–vis region have been used for quantitating anionic surfactants (32), barium (33), boron (34), iron (35), sulfate (36), sulfide (37), and total petroleum hydrocarbons (TPH) (38). Anionic surfactants can be monitored near 655 nm as a complex with methylene blue according to EPA Method 425.1. Chloride solutions have been shown to give false positives with this approach (39). Barium has been measured as low as 2 mg/L as a precipitate after adding a sodium sulfate mixture and quantified at 450 nm. Strontium, silica, and calcium are the most detrimental interferences that may be encountered using this approach. Boron is measured at levels above 2 mg/L as the reaction product with carmine at 605 nm. Iron is monitored with the colorimetric phenanthroline indicator at 510 nm after the reagent has converted most forms of iron to a soluble ferrous iron. Iron can be measured down to 0.1 mg/L with the most common interference being a cumulative concentration of Ba2+ and Sr2+ greater than 50 mg/L. Sulfate at levels greater than 2 mg/L can be measured at 450 nm through a turbidimetric method after precipitation as barium sulfate. However, barium, magnesium, and silica present in the water sample can interfere with the accuracy of these results. Sulfide can be detected spectroscopically down to 0.01 mg/L at 665 nm after reacting with N,N-dimethyl-p-phenylenediamine sulfate to form methylene blue. A semiquantitative method for TPH, which is a cumulative measurement of hydrocarbons ranging from C6 to C36, uses an immunoassay in which the hydrocarbons and enzyme compete to bind to antibodies immobilized on the cuvette. Measuring this absorbance at 450 nm yields a sensitivity equivalent to at least 2 mg/L diesel fuel. Chlorine present in solution can interfere with the assay.
TPH can also be measured by IR spectroscopy. Previously, a method consisting of serial extractions with fluorocarbon-113 (CFC-113) and silica drying has been shown to be able to generate an extract adequate to quantify the absorbance of C-H stretches at 2950 cm-1. Since CFC-113 is an ozone-depleting substance, the EPA has discontinued the method and suggests using the ASTM International Method D7006-04, which uses S-316 as a CFC substitute.
OES has become the chosen technique for measuring dissolved metals in UOGWW (2). Metals of interest such as Ba, Sr, Fe, Na, Ca, and Mg are easily in the milligram-per-liter concentration range, comfortably above detection limits for inductively coupled plasma (ICP)-OES. The alkali and alkaline earth metals can be at levels of hundreds to thousands of milligrams per liter depending on the contribution of formation water to the overall UOGWW mixture. These excessive concentrations can be detrimental to an ICP-MS, accepted to be more sensitive than the ICP-OES. Most ICP-OES instruments come with the option to change between axial and radial viewing modes to assist in measuring samples across a wide concentration range, resulting in a much wider linear range for quantification than ICP-MS (40). Great attention needs to be taken in wavelength selection for the ICP-OES to ensure there is no spectral overlap from other metals at high concentrations or unexpected interferences from other hydraulic fracturing additives. Atomic absorption could also be implemented, but lacks the multielement throughput of ICP-OES.
The majority of research and application notes involving metals analysis with ICP-OES have been toward profiling UOGWW. These samples are currently a national disposal issue, a challenging matrix to overcome when making measurements, and possess a set of inorganic “known knowns” like brine salts to target. Our more recent investigation (27) of water quality overlying the Barnett Shale used ICP-OES (ICPE-9000 from Shimadzu Scientific Instruments, Inc.) for measurement of 13 metals most relevant to UOG exploration and that exhibited minimal spectral interferences. Strontium was the only metal determined to be above the 4.0 mg/L maximum contaminant level (MCL) by ICP-OES. Standard addition was used for quantification to overcome unpredictable matrix effects that had previously been observed (41).
ICP-MS is another approach for elemental analysis, primarily for trace metals in water. The MS detector is more sensitive than using OES, but has a limited dynamic range. Our groundwater studies (26,27) have used ICP-MS (Varian 820 ICP-MS) for the quantification of arsenic and selenium. Arsenic with an MCL of 10 µg/L and selenium at 50 µg/L warrant the additional sensitivity of ICP-MS for adequate quantitation. Strontium and barium were also measured by ICP-MS in 2011 because of instrument availability (26). In 2011, As, Se, and Sr each were shown to have a negative correlation with proximity to UOG wells (that is, higher values in water wells closer to UOG wells). The most plausible conclusion was that an increased pH and mechanical vibrations from the neighboring UOG activity liberated iron oxide that had complexed these metals in poorly maintained water wells. In 2011, 29 of the 100 samples exceeded the EPA MCL for arsenic, but only 10 of the 550 samples in 2014 exceeded the limit. It is hypothesized that the reduction in UOG exploration between the sampling campaigns reduced subsurface vibrations, in turn reducing the amount of dissolved arsenic. The water quality in 2014 was also found to be a less reducing environment, which would decrease the solubility of arsenic in groundwater.
Concluding Remarks
Significant efforts have been made in this decade, by predominately academic institutions, to understand the environmental, social, and economic effects of UOG exploration. The collaborations that have formed through this multifaceted research have generated astounding conclusions to date, but nearly all have commenced without technical or chemical advice from industrial partners. The guidance from drilling operators would allow for more focused and efficient analytical methods and more effective conclusions, which could in turn ease the public opinion of UOG. The detection of most of these aforementioned compounds can occur in groundwater through avenues other than UOG, convoluting the ability to identify the source. The burden has been on the researcher to present exhaustive evidence if contamination from UOG has been suggested, but operators are able to merely discredit the research and rely on the uncertainty of these other possibilities. It is expected that overcoming this hurdle will only happen once proprietary chemicals or tracers are incorporated, so that contamination events can be clearly attributed to a particular UOG process and operation. Until then, conclusions will continue to be deduced through disproving all other possibilities, which is not an efficient route to take.
References
(1) S. Stein, Anal. Chem.84, 7274–7282 (2012).
(2) D.D. Carlton Jr., Z.L. Hildenbrand, B.E. Fontenot, and K.A. Schug, in Hydraulic Fracturing Impacts and Technologies, V. Uddameri, A. Morse, and K.J. Tindle, Eds. (Taylor & Francis Group, New York, 2016), pp. 115–132.
(3) S.G. Osborn, A. Vengosh, N.R. Warner, and R.B. Jackson, Proc. Natl. Acad. Sci. U.S.A. 108, 8172–8176 (2011).
(4) R.E. Jackson, A.W. Gorody, B. Mayer, J.W. Roy, M.C. Ryan, and D.R. Van Stempvoort, Ground Water51, 488–510 (2013).
(5) A. Kissinger, R. Helmig, A. Ebigbo, H. Class, T. Lange, M. Sauter, M. Heitfeld, J. Klünker, and W. Jahnke, Environ. Earth Sci.70, 3855–3873 (2013).
(6) D.A. Stolper, M. Lawson, C.L. Davis, A.A. Fereira, E.V. Santos Neto, G.S. Ellis, M.D. Lewan, A.M. Martini, Y. Tang, M. Schoell, A.L. Sessions, and J.M. Eiler, Science344, 1500–1503 (2014).
(7) S.G. Osborn and J.C. McIntosh, Applied Geochemistry25, 456–471 (2010).
(8) R.B. Jackson, A. Vengosh, T.H. Darrah, N.R. Warner, A. Down, R.J. Poreda, S.G. Osborn, K. Zhao, and J.D. Karr, Proc. Natl. Acad. Sci. U.S.A.110, 11250–11255 (2013).
(9) M. Schoell, Geochim. Cosmochim. Acta44, 649–661 (1980).
(10) L. Chambers, OI Analytical, Application Note 37920312 (2012).
(11) F. Hudson, US EPA, RSK SOP-175 (2004).
(12) Z. Ji, Agilent Technologies, Application Note 228–387 (2000).
(13) Isotech Laboratories, Collection of Groundwater Samples from Domestic and Municipal Water Wells for Dissolved Gas Analysis http://www.isotechlabs.com/customersupport/samplingprocedures/IsoBagSM.pdf (August 6, 2014) (2011).
(14) K.A. Schug, I. Sawicki, D.D. Carlton Jr., H. Fan, H.M. McNair, J.P. Nimmo, P. Kroll, J. Smuts, P. Walsh, and D. Harrison, Anal. Chem.86, 8329–8335 (2014).
(15) L. Bai, J. Smuts, P. Walsh, H. Fan, Z. Hildenbrand, D. Wong, D. Wetz, and K.A. Schug, J. Chromatogr. A 1388, 244–250 (2015).
(16) Analysis of Hydraulic Fracturing Fluid Data from the FracFocus Chemical Disclosure Registry 1.0, (U.S. Environmental Protection Agency, Washington, D.C., EPA/601/R-14/003, i-155, 2015).
(17) R.B. Jackson, E.R. Lowry, A. Pickle, M. Kang, D. DiGiulio, and K. Zhao, Environ. Sci. Technol.49, 8969–8976 (2015).
(18) A. Vengosh, R.B. Jackson, N.R. Warner, T.H. Darrah, and A. Kondash, Environ. Sci. Technol.48, 8334–8348 (2014).
(19) C. Brufatto, J. Cochran, L. Conn, and D. Ower, Oilfield Review Autumn, 62–76 (2003).
(20) R.D. Vidic, S.L. Brantley, J.M. Vandenbossche, D. Yoxtheimer, and J.D. Abad, Science 340, 1235009 (2013).
(21) A.R. Ingraffea, M.T. Wells, R.L. Santoro, and S.B.C. Shonkoff, Proc. Natl. Acad. Sci. U.S.A.111, 10955–10960 (2014).
(22) United States House of Representatives Committee on Energy and Commerce,
, (2011).
(23) www.fracfocus.org/chemical-use (April 1, 2014).
(24) D.C. DiGiulio, R.T. Wilkin, C. Miller, and G. Oberley, Investigation of Ground Water Contamination Near Pavillion, Wyoming, (U.S. Environmental Protection Agency, Washington, D.C., 2011).
(25) Shimadzu Europa GmbH, Linear Retention Index Function (LRI) in GCMSsolution 2.4 http://www2.shimadzu.com/applications/gcms/Appl_GCMS_Ret-index_07C_066_en.pdf (January 23, 2013).
(26) B.E. Fontenot, L.R. Hunt, Z.L. Hildenbrand, D.D. Carlton Jr., H. Oka, J.L. Walton, D. Hopkins, A. Osorio, B. Bjorndal, Q.H. Hu, and K.A. Schug, Environ. Sci. Technol.47, 10032–10040 (2013).
(27) Z.L. Hildenbrand, D.D. Carlton Jr., B.E. Fontenot, J.M. Meik, J.L. Walton, J.T. Taylor, J.B. Thacker, S. Korlie, C.P. Shelor, D. Henderson, A.F. Kadjo, C.E. Roelke, P.F. Hudak, T. Burton, H.S. Rifai, and K.A. Schug, Environ. Sci. Technol. 49, 8254–8262 (2015).
(28) T.D. Hayes and B.F. Severin, “Characterization of Flowback Waters from the Marcellus and Barnett,” Gas Technology Institute (2012).
(29) T. Colborn, K. Schultz, L. Herrick, and C. Kwiatkowski, Hum. Ecol. Risk Assess. 20, 86–105 (2014).
(30) S.K. Ritter, C&EN92, 31–33 (2014).
(31) Hach Company, Hydraulic Fracturing Water Analysis Handbook,www.hach.com/asset-get.download.jsa?id=17255280808 (July 29, 2014) (2013).
(32) P.R. Wright, P.B. McMahon, D.K. Mueller, and M.L. Clark, U. S. Geological Survey, Data Series 718, (2012).
(33) Hach Company, Barium, Turbidimetric Method 10251 www.hach.com/asset-get.download.jsa?id=9595814105 (July 29, 2014).
(34) Hach Company, Boron Carmine Method 10252 www.hach.com/asset-get.download.jsa?id=9595814103 (July 29, 2014).
(35) Hach Company, Iron, Total Method 10249 www.hach.com/asset-get.download.jsa?id=9595814104 (July 29, 2014).
(36) Hach Company, Sulfate Method 10248 www.hach.com/asset-get.download.jsa?id=9595814106 (July 29, 2014).
(37) Hach Company, Sulfide Methylene Blue Method 10254 www.hach.com/asset-get.download.jsa?id=11122402601 (July 29, 2014).
(38) Hach Company, TPH (Total Petroleum Hydrocarbons) Immunoassay Method 10050 www.hach.com/asset-get.download.jsa?id=7639983907 (July 29, 2014).
(39) A.L. George and G.F. White, Environ. Toxicol. Chem. 18, 2232–2236 (1999).
(40) PerkinElmer Inc., “Atomic Spectroscopy - A Guide to Selecting the Appropriate Technique and System,” http://www.perkinelmer.com/CMSResources/Images/44-74482BRO_WorldLeaderAAICPMSICPMS.pdf (September 10, 2013) (2013).
(41) D.D. Carlton Jr., B.E. Fontenot, Z.L. Hildenbrand, T.M. Davis, J.L. Walton, and K.A. Schug, Int. J. Environ. Sci. Technol. April, 1–10 (2015).
Doug D. Carlton Jr., and Kevin A. Schug are with the Department of Chemistry and Biochemistry and the Collaborative Laboratories for Environmental Analysis and Remediation at The University of Texas at Arlington in Arlington, Texas. Zacariah L. Hildenbrand is with the Collaborative Laboratories for Environmental Analysis and Remediation at The University of Texas at Arlington and Inform Environmental, LLC, in Dallas, Texas. Direct correspondence to: kschug@uta.edu

















Best of the Week: AI and IoT for Pollution Monitoring, High Speed Laser MS
April 25th 2025Top articles published this week include a preview of our upcoming content series for National Space Day, a news story about air quality monitoring, and an announcement from Metrohm about their new Midwest office.




LIBS Illuminates the Hidden Health Risks of Indoor Welding and Soldering
April 23rd 2025A new dual-spectroscopy approach reveals real-time pollution threats in indoor workspaces. Chinese researchers have pioneered the use of laser-induced breakdown spectroscopy (LIBS) and aerosol mass spectrometry to uncover and monitor harmful heavy metal and dust emissions from soldering and welding in real-time. These complementary tools offer a fast, accurate means to evaluate air quality threats in industrial and indoor environments—where people spend most of their time.


