Applications of Raman Spectroscopy in Solvent Distillation and Exchange During Early-Phase Chemical Synthesis
Applications of Raman spectroscopy in monitoring the concentration of solvents in various distillation and solvent exchange steps in chemical synthesis are discussed. Two case studies from early-phase active pharmaceutical ingredient (API) process development, one each from the distillation and the solvent exchange operations, are presented. The results are compared to respective conventional techniques. Sampling, measuring, and building models using appropriate chemometric tools are described in detail. A list of 70 different pairs of commonly used solvents and reactants, where Raman spectroscopy models could be successfully developed and employed, is provided with the corresponding range of quantitation. The advantages of Raman spectroscopy, such as rapid and nondestructive analysis, suitability for process analytical technology (PAT)-based applications for real-time monitoring, and ease of automation, are highlighted over traditional approaches. Some practical challenges of the technique towards its implementation are also discussed.
Raman spectroscopy can be an invaluable analytical tool for qualitative and quantitative applications and molecular fingerprinting. The technique has been widely explored for various applications in recent years. Nondestructive analysis, quick data acquisition times, and spectra rich with baseline resolvable features are among the most encouraging aspects of this technique. With the development of Raman probes with enduring compatibility, PAT also finds applications for usage in hazardous reactions without offline sampling (1). With early applications focused on photonics (2) and petroleum products (3), Raman PAT tools were successfully extended to several other fields such as synthetic chemistry (4,5), material science (6), medicine (7,8), and forensics (9).
The pharmaceutical industry has mostly relied on a conventional offline mode of analysis because of a high level of regulatory and compliance requirements. Raman spectroscopy was largely limited to solid form identification testing using a Fourier transform Raman spectrometer (10). However, with an increasing demand for large-scale drug product manufacturing with improved turnaround time, the potential for real-time analysis with appropriate quality controls was gradually explored. In 2004, the U.S. Food and Drug Administration (FDA) came up with industry-wide guidance for utilizing PAT tools in pharmaceutical manufacturing (11). This guidance opened the possibility of implementing atline and inline testing of samples and increased the scope of Raman spectroscopy-based tools in regulated manufacturing plants (12). The wealth of data generated with Raman PAT tools provided detailed insights into the process, facilitating the usage of design of experiment (DoE) and quality by design (QbD) approaches with the help of chemometric tools (13). Figure 1 shows the number of publications in Raman spectroscopic analysis relevant to the pharmaceutical industry in the last two decades. A significant proportion of the reports highlight its use as a PAT tool, suggesting increased applications of this technique in modern pharmaceutical development.


FIGURE 1: The number of publications per year in Raman spectroscopy applications to pharmaceuticals for 2004–2020. The solid top section of each bar represents the number of publications highlighting the application of Raman spectroscopy as a PAT tool, reported in that year.
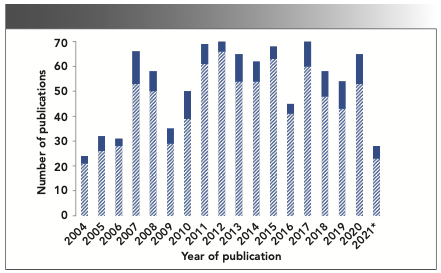
The publications mentioned in Figure 1 broadly cover almost all the critical unit processes or operations of small molecule API manufacturing and oral solid dosages (OSD) development, such as reaction monitoring (5,14), polymorphic transformations (15), crystallization (16), blend uniformity (17,18), and bioprocessing (19,20). Although Raman-based PAT tools’ utility for tracking reaction intermediates and products is well studied (21,22), its utility for solvent content determination, a step common to almost every synthetic procedure, remains significantly underexplored.
Chromatographic techniques, like gas chromatography (GC) and liquid chromatography (LC), are commonly utilized to measure solvent concentrations and to further understand the reaction process. PAT tools offer several advantages. They are versatile and can be employed as an atline, inline, or offline tool in a process development laboratory. In addition, they provide a quick qualitative monitoring of reactants or solvents or both. They can also use chemometrics, which makes it an attractive option for performing quantitative analysis (23), and the model from early development trials can be extended to scale-up commercial manufacturing with minimal bridging work.
Although a few publications are exploring the use of near-infrared (NIR) spectroscopy (24–26) and mass spectrometry (27,28) for monitoring solvents during distillation, Raman-based PAT method development reports in this area are low. In general, NIR seems to work well for a variety of solvents. However, its applicability to a wider class of solvents is limited because of a lower degree of analyte specificity (29) and difficulty in building NIR quantitative models. On the other hand, mass spectrometric tools rely on the corresponding vapor phase analysis. Therefore, they do not serve a complete purpose as a PAT tool for direct inline measurements (30). In a recent article by Wasylyk and others (31), a Raman spectroscopy-based PAT method was introduced as an equivalent alternative to the GC method for the quantitative estimation of solvents in a solvent swap (exchange) process on a commercial manufacturing scale. Successful demonstration of an online measurement for a pharmaceutical compound using full-fledged Raman probes with equipped reactors assured the feasibility of such a setup under a highly regulated environment.
Along with specific advantages discussed earlier, applying Raman spectroscopic methods at the early stages of API manufacturing can help ensure a robust analytical development strategy. Its exploration at the early stages of method development could facilitate the readiness of methods for large-scale manufacturing campaigns, accelerate the development timelines of pharmaceutical assets, and promote continuous manufacturing through the online monitoring of reactions. This article presents two case studies, one each for the distillation and the solvent exchange operation steps, from the early phase API synthesis projects at Bristol Myers Squibb. The method development and building of chemometric quantitative models are discussed in detail, verifying the results with a conventional GC technique.
Materials and Methods
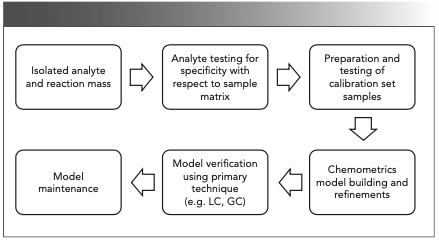
Quantitative analysis using Raman spectroscopic techniques involves various steps, which are outlined in Figure 2, and the experimental details for each step are discussed in the following subsections.


FIGURE 2: Steps involved in quantitative analysis using Raman spectroscopy as a PAT tool.
Materials
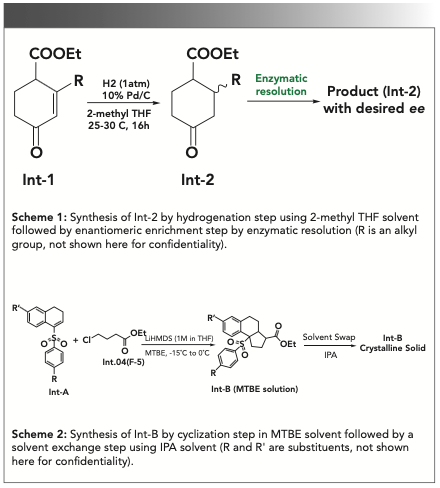
All solvents used in the reaction and for model development were commercially available, and the potency of solvents was considered 100%. GC grade acetonitrile and isopropyl alcohol were purchased from Sigma-Aldrich. The materials and products mentioned in schemes 1 and 2 (Figure 6) were synthesized in-house, and their chemical structures have not been revealed for confidentiality reasons.


FIGURE 6: Synthetic Schemes 1 and 2 for the discussed case studies.
Instruments and Methods
Raman Spectroscopy
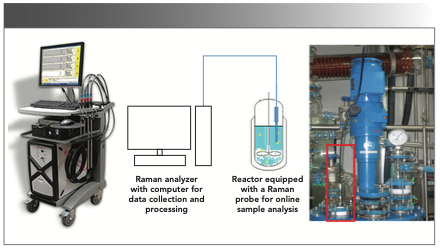
The Raman measurements were performed with a Kaiser Raman RXN2, using an Invictus 785-nm NIR diode laser, with a spectral coverage of 150–3425 cm-1 and operating with 400 mW of power. The system was equipped with a low noise charge-coupled device (CCD) detector (−40 ̊C) to avoid the dark noise and baseline offset. A laser was coupled to the Kaiser Mk II filtered fiber optic probe head, and it was placed in an analytical sample chamber or coupled to a wet head immersion probe using a fiber optic cable. A probe that was 0.5 inches in diameter and 18 inches in length (made of stainless steel with sapphire window) with a short fixed-focus was used for inline analysis. This probe complied with Atmosphères Explosibles (ATEX) standards. Instrument calibration was performed for the spectrograph wavelength, the laser wavelength, and the laser intensity. Its performance was verified using cyclohexane for the peak location and intensity. While performing distillation or solvent exchange, the probe was immersed into the reaction mass under constant agitation, or stirring, conditions. Before each analysis, dark spectrum subtraction and a cosmic ray filter were applied. The atline analysis sample was placed in an analytical sample compartment—a closed system with an automatic interlock that prevents stray light. Glass (reaction vessels) was covered with glass wool and aluminum, preventing stray light. Also, the software had options set for dark backgrounds if there was concern about stray light during the experiments. Raman data was collected using the HoloReact software package by Kaiser Optical Systems Inc. Figure 3 provides a schematic view of inline measurement using a Raman analyzer and the corresponding actual setup used in the author’s laboratory.


FIGURE 3: Schematic diagram of the experimental setup used for inline sample analysis from the reactor equipped with a Raman probe. Photographs in the extreme left and right show the view of a Raman analyzer and a reactor in the authors’ laboratory.
Gas Chromatography
An Agilent gas chromatograph (model 7890A), equipped with the 7683 series autosampler and 7683B series injector, was used for the analysis. For monitoring solvent content in case study-1, aDB-1 column (30 m x 0.53 mm i.d. x1.5 μm film thickness) was used with nitrogen as the carrier gas. A flame ionization detector (FID) was used for detection with a temperature set at 300 °C. Then, 1 mL of the sample (neat injection) was injected into the split port maintained at 200 °C with a constant flow rate of 4 mL/min (constant flow mode). The split ratio was 10:1, and the oven temperature was maintained at 40 °C for 4 min and increased at a rate of 15 °C/min to 150 °C before being held for 1 min and followed by a further increase at the rate of 30 °C/min to 280 °C, and then held for 8.33 min with a runtime of 25 min. Acetonitrile was used as a needle wash solvent.
In case study-2, a DB-624 column (75 m x 0.53 mm i.d. x 3.0 μm film thickness) was used with nitrogen as a carrier gas, and the detector temperature was set at 260 °C. First, 1 mL of a sample (neat injection) was injected into the split port maintained at 175 °C with a flow rate of 4 mL/min (constant flow mode). The split ratio was 20:1, and the oven temperature was maintained at 100 °C for 7 min before being increased at a rate of 30 °C/min to 240 °C, and held for 8.33 min with a runtime of 20 min. The needle wash was performed using isopropyl alcohol.
Preparation of Calibration Standards
A set of calibration samples was generated to build a quantitative Raman model to determine respective solvents in studies 1 and 2.
In case study 1, calculated amounts of 2-methyl tetrahydrofuran (THF) were spiked in the isolated product from the hydrogenation step (reaction mass with a known amount of 2-methyl THF) to generate the standard samples with solvent levels ranging from 1–10% (Table Ia). GC was used to analyze each sample to obtain the reference values.


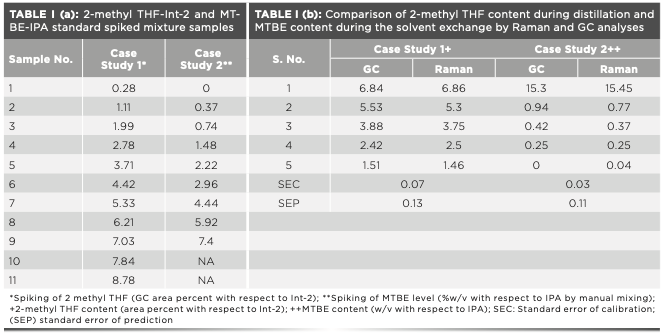
In case study 2, the solvent mixtures of methyl tert-butyl ether (MTBE) and isopropyl alcohol (IPA) were prepared in the range of 0.5–10% v/v, corresponding to 0.37–7.4% w/v of MTBE in IPA as shown in Table Ia.
Raman Model Development
For the solvent systems to be studied, different ratios of solvent combinations were prepared, and GC was used to analyze the standards to obtain the reference set values. Raman models were then built by constructing calibration set samples, using data points from the Raman and GC measurements. The relative concentration of solvents in the standard samples and the range of the calibration set was based on the quantification levels required for the process. The accumulation times were adjusted by modifying the laser exposure time in seconds and the number of accumulations (number of repetitions of the scan) so that the saturation of CCD was avoided. Wherever the compound exhibited fluorescence, exposure times were decreased, and the optimum signal intensity was achieved by increasing the number of accumulations. A multivariate chemometrics model was built for each case to analyze test samples. During model building, each solvent ratio of the calibration set was collected by an “atline” sample chamber for a minimum of six counts (number of complete acquisition cycles) to account for the natural variance in the intensity of the Raman spectrum. Once the acquisitions for all the samples with varying solvent ratios were collected, distinguishing peaks for each solvent were identified to ensure that the model was specific for that solvent. These peaks were selected to have minimum interference with other matrix analytes in the reaction. The solvent to be quantified was referenced either with one of the analytes of interest or with the exchanged solvent.
Furthermore, the ratio of the peak intensities of the analyte and the reference were used so that peak intensities could be normalized. The proportional relationship between the Raman scattering intensity and analyte concentration (weight fraction or concentration of solvent) was the basis for the quantitative analysis done in this study. The accuracy of the model was evaluated by the statistical parameter standard error of calibration (SEC) as per equation (1) (32,33):


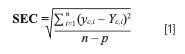
where yC = the Raman predicted value obtained from calibration set sample; YC = the reference value obtained from same calibration set value; n = number of samples; and p = number of coefficients in the model.
The predictive capacity of the model was also measured by calculating the statistical parameter standard error of prediction (SEP) as per equation (2)


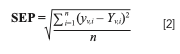
where yV = the Raman predicted value; YV = the reference method value; and n = number of samples.
Once a model was developed, it was subject to the stability test wherein the software performed a “thermal-shock” test of the model. The stability test modifies each weighting factor in the model and attempts to recalibrate the model until a true minimum is reached. Once the model was finalized, the solvent content was determined by collecting the Raman spectra for the samples of interest.
Results and Discussion
This study explored the utilization of Raman-based PAT tools to determine solvent content during distillation and monitor solvent “exchanges” or exchanges in the laboratory-scale reactors. Two case studies are discussed where the level of solvent impacted the product quality, product yield, and the effect of the residual solvent in the next step. Raman models were built for the selected systems by analyzing calibration set samples. Different ratios of solvent or solvent combinations were prepared. The samples were analyzed using GC to obtain the reference set values. Once the model performance was established, the model was applied to estimate the solvent content during inline distillation and solvent exchange.
Case Study 1: Inline Solvent Content Determination during Distillation
During the synthesis of an API under clinical investigation, a key step was the hydrogenation of cyclohexene intermediate (Int-1) to obtain an advanced intermediate (Int-2), followed by enzymatic resolution and purification to get the product with a desired chiral purity (Scheme 1 in Figure 6). In an enzymatic resolution, solvents have a significant impact, and it is critical to monitor and control solvent composition as necessary (34,35). During the optimization of the synthetic process, it was observed that the residual 2-methyl THF content present in the hydrogenation step was significantly impacting the enzymatic resolution step. The higher the 2-methyl THF content during the enzymatic resolution step, the lower was the enantiomeric excess (that is, the yield of the product).
The enzymatic resolution, an important step in this synthesis to enrich the desired enantiomer as the undesired isomer, contributed to higher impurity at the API step. Purification to remove the undesired enantiomer at subsequent steps led to significant material loss. Hence, controlling the 2-methyl THF content in the hydrogenation step was important. As per the solvent content (by GC) data on the laboratory trial batches, chiral purity was found to vary between 88–95% for the 2-methyl THF content of 12 to 3 area percentage (AP) relative to the product (Int-2) peak, respectively. Importantly, obtaining a solvent-free product was practically challenging as the compound (Int-2) exists in the liquid state at room temperature and was completely miscible with the solvent 2-methyl THF. Furthermore, the enzymatic resolution was working well up to 3% residual amounts of 2-methyl THF without compromising the desired chiral purity of the product. Accordingly, a limit of not more than 3% of 2-methyl THF was fixed for the scale-up batches.
Raman spectra for all the components in the reaction (solvent, starting material, and the product) were recorded to evaluate the feasibility of Raman spectroscopic analysis for the 2-methyl THF content determination in the reaction mass. It was noted that a peak at 905–930 cm-1 emerged from the analyte of interest; that is, 2-methyl THF had no significant interference with the other components tested and could potentially be used as a unique peak to track and measure its content in the reaction mass. Moreover, a peak from the product (Int-2) at 720–771 cm-1 was iden- tified that could be used to calculate the peak ratio (I2MeTHF/IInt-2 + I2MeTHF) required for the quantitative analysis across the samples. Therefore, based on the preliminary specificity evaluations, it was clear that Raman spectroscopic analysis can monitor the content of 2-methyl THF in the reaction mass.
As described in the experimental section, calibration samples were generated to build a quantitative Raman model to determine 2-methyl THF content in the reaction mass. These samples were analyzed by Raman spectroscopy, and a multivariate calibration model was built by keying in corresponding results from GC. The exposure was 3.5 s for 15 accumulations with a CCD saturation value (level of saturation of CCD detector) of 30, and each sample was scanned for six counts. The 2-methyl THF peak was integrated at 905–930 cm-1 and the product peak was at 720–771 cm-1 for the Raman shift. Although there was slight interference from the starting material in the product region, it was considered insignificant because the amount of starting material before the distillation of the product was approximately 1%, which would have a minimal impact on the model predictability. Also, the effects of baseline correction were evaluated, and the models built with Pearson’s baseline correction and without baseline correction were comparable (SEC observed for both the models were approximately 0.07).
After getting the quantitative Raman model in place, the inline Raman data were collected for a scale-up batch (batch size = 17.0 kg) performed in a reactor fitted with the Raman probe. The sample in the reactor was scanned at the time interval of 15 minutes during the solvent distillation step (45 °C under vacuum). The exposure was 3.0 s for five accumulations with a CCD saturation value of 30. The data was processed, and the 2-methyl THF content as AP at various time points was predicted from the model. Figure 4 shows the plot of 2-methyl THF content in the reaction mass as a function of time. The overlay of the Raman spectra (650–1000 cm-1) for the individual scans collected is shown in Figure 4.

. The inset in Figure 4 shows the Raman spectra overlay of different time points samples for distillation of 2-methyl THF from the product (Int-2).")
FIGURE 4: Inline measurement of 2-methyl THF content during the distillation of 2-methyl THF from the product (Int-2). The inset in Figure 4 shows the Raman spectra overlay of different time points samples for distillation of 2-methyl THF from the product (Int-2).
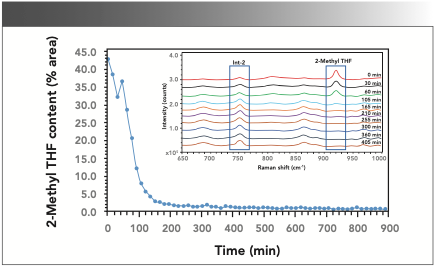
A steep decline in the content of 2-methyl THF was noted at the beginning of the distillation process (reflux heat at 45 °C under vacuum). Although the rate of decrease was quite high in the first two hours, it seemed to decrease slowly during the next hour and almost attained minimum concentration after that. The uncharacteristic behavior seen around the third and fourth-time points could be either because of sample nonhomogeneity or it could be an artifact. Interestingly, the inline analysis indicated that distillation could be completed in 3 h, whereas a period of 4 h was typically reserved for this distillation process. Based on this inline Raman data, it was justified that the distillation could be stopped after 3 h, potentially saving 25% of the reactor time for this step.
Also, the accuracy of the 2-methyl THF content predicted from the Raman model was verified by performing GC analysis for a set of five samples containing different amounts of 2-methyl THF. Notably, the solvent content determined from GC analysis was predicted using the Raman model and was in excellent agreement with a SEP of 0.13, confirming the suitability of Raman spectroscopy-based PAT tools for such measurements (Table Ib).
Case Study-2: Inline Solvent Content Determination During Solvent Swap or Exchange
During the synthesis of an intermediate, the reaction solvent methyl tertbutyl ether (MTBE) was required to be exchanged by isopropyl alcohol (IPA). This exchange of solvents was necessary to improve the yield of the product because the product had a relatively higher solubility in MTBE, and good precipitation was observed in IPA (Scheme 2 in Figure 6). With optimized reaction conditions, the limit for MTBE content was set at NMT 1.0 % w/v, with respect to IPA, balancing the desired yield versus reaction time.
With both the solvents, MTBE, and IPA having strong Raman signals, the application of the Raman-based PAT tools for monitoring this solvent exchange reaction was explored. The model development was carried out by keying the solvent concentrations known from the preparation of standards. Spectral regions of noninterfering peaks from MTBE (711–736 cm-1) and IPA (800–836 cm-1) were selected for creating the model. The exposure was 5 s for 10 accumulations with a CCD saturation value of 33, and each sample was scanned for six counts. No preprocessing was applied, and both peaks were baseline integrated with a baseline order of 1 and baseline points of 1. A multivariate calibration curve was developed with an SEC of 0.03. The model was independently verified by testing some known composition mixtures of MTBE and IPA at different concentration levels.
After the successful model building, inline Raman data were collected for a scale-up batch (batch size = 0.8 kg) performed in a reactor fitted with the Raman probe. The reaction mass was scanned at an interval of 15 min during the solvent exchange step with an exposure time of 2.0 s (10 accumulations). The overlay of Raman spectra (200–1000 cm-1) for the individual scans collected is shown in Figure 5 inset.


FIGURE 5: Inline monitoring of MTBE content during the solvent exchange reaction of MTBE with IPA. The inset in Figure 5 shows the Raman spectra overlay of different time point samples during the solvent exchange of MTBE with IPA.
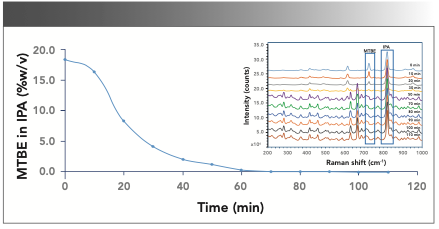
The MTBE content predicted by the inline Raman analysis at various time points indicated a decrease in concentration over time, as shown in Figure 5. Notably, the inline analysis indicated that distillation could be completed in 1.5 h, compared to the planned 4 h time per the batch manufacturing records, potentially saving 2.5 reactor hours per step.
Some samples were also analyzed by the GC technique to confirm the accuracy of readings obtained by inline Raman analysis. Table Ib shows that the solvent content determined by Raman and GC methods agree with a SEP of 0.11.
Although Raman spectroscopic analysis as a PAT tool provides several advantages over conventional approaches, some of the practical challenges limit the use of this methodology. Potential interference from the reaction matrix is one of the most common challenges encountered during model development for quantitative analysis using the Raman PAT method. Although adequate specificity can be obtained in most cases, the reaction intermediates and products can significantly interfere with the solvent peak of interest. Also, the extent of matrix interference can vary during solvent distillation and exchange because of phase changes resulting from solubilization or precipitation of products. In such cases, quanti- tative measurement using Raman methods can be challenging, and the results derived from a quantitative model may not be accurate. Hence, random checks with a primary reference technique such as GC may be necessary as a part of model performance verification.
Quantitative Raman Models for Solvent Exchange or Exchange Operations
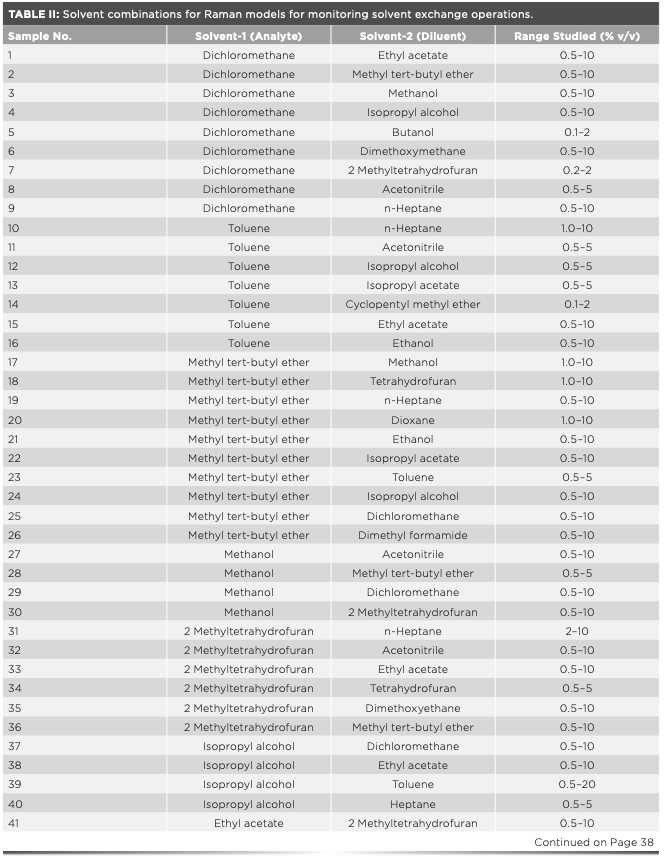
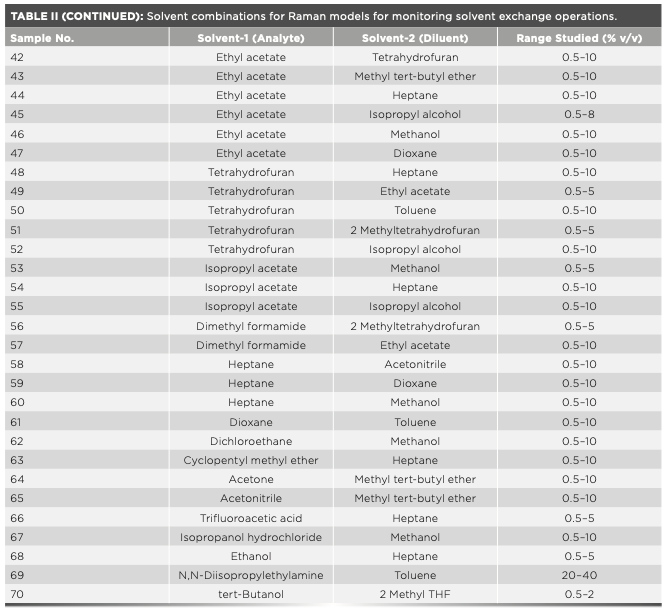
Solvent exchange is one of the most common activities during API synthesis, isolation, and crystallization. Depending on the need and suitability, various solvents are used based on parameters such as solubilizing potential, boiling point, polarity, viscosity, and toxicity. These solvents possess a characteristic Raman spectral signature and exhibit reasonable to high Raman response because of their different chemical structures. Encouraged by some early results from successful inline and offline solvent exchange monitoring using Raman spectroscopy, a library of quantitative Raman models for solvent pairs used commonly during API synthesis was built. Table II provides the list of solvent combinations where Raman-based PAT tools have been successfully employed for monitoring solvent exchange operations from some of our early-phase API synthesis projects. Table II also provides a lower range of quantification tested in each case using an optimized experimental setup as per the respective project requirements.




Raman Model Transferability
Importantly, once developed, the model for a particular pair of solvents (or a pair of solvent and product) can be extended for other reactions and operations with similar solvent content monitoring needs. Potential interference from the reaction mass (for example, starting materials, products, intermediates, and impurities) is one of the major challenges in deciding the suitability of Raman methods from one reaction to the other. A simple feasibility test by recording Raman spectra of a reaction mass in the spectral range of interest confirms the extent of interference and applicability of the existing model to the concerned reaction. In our experience, the solvent swap and exchange model, once developed, shows high transferability from reaction to reaction involving various compounds. Also, with the necessary infrastructure (Raman probes of suitable dimensions), the same models can be used for inline analysis and offline analysis from small-scale research and development trials.
Conclusion
With the help of ever-evolving chemometrics tools, the wealth of information collected through inline Raman spectroscopic analysis can be utilized to gain increased process understanding. This advancement in chemometrics and data collection also facilitates the implementation of modern control strategies through DoE and QbD while satisfying the immediate intended analytical purpose of the method. The case studies discussed here successfully demonstrate the use of inline Raman spectroscopic analysis to estimate solvent content during the solvent exchange and distillation operations. Simple multivariate chemometric models were developed by integrating the peaks of the solvent of interest and product or another solvent to monitor the residual solvent content during distillation and solvent exchange steps. Raman spectroscopy provided a quick estimation of levels of solvents to understand the progress of distillation (solvent) exchange without waiting for time-consuming offline analysis. It saves reactor time for a batch and facilitates an improved process understanding. Reliability and robustness of the quantitative measurements by Raman methods are demonstrated through verification of solvent content measurements by the reference technique (GC). A list of 70 pairs of commonly used solvents where Raman models were established with the corresponding range of quantitation values is published. This information would serve as a good starting point to understand the feasibility and scope of Raman methods for solvent content determinations of interest.
Acknowledgments
The authors thank Lakshmikant Bajpai of Analytical R&D at BBRC for valuable suggestions and manuscript review. Our sincere thanks to Ming Huang, John Wasylyk, Joel Young, and Jay Brumfield of Bristol Myers Squibb for their support of this work. The authors also acknowledge Syngene International Limited for providing the infrastructure to perform this work and chemical development and API supply at BBRC for providing samples used in this work.
References
(1) “Raman Spectroscopy – A Tutorial,” Kaiser Optical Systems, Inc, an Endress+Hauser Company.
(2) J. Popp, B. Dietzek, M. Schmitt, P. Rösch, R. Möller, and C. Krafft, “Raman spectroscopy – An essential tool for bio photonics,” paper presented at the International Workshop on Biophotonics, Parma, Italy, 2011.
(3) D. Orange, E. Knitile, D. Farber, and Q. Williams, in Mineral Spectroscopy, M.D. Dyar, C. McCammon, and M.W. Scaefer, Ed. (Geochemical Society Special Publications, Washington, D.C., 1996), vol. 5, pp. 65–81.
(4) D. Cohen, A. Cosbie, R. Milburn, S. Shaw, and Y. Xie, Org. Proc. Res. Dev. 18, 1807–1811 (2014).
(5) I. Csontos, H. Pataki, A. Farkas, H. Bata, B. Vajna, Z.K. Nagy, et al, Org. Proc. Res. Dev. 19, 189–195 (2015).
(6) Y. Liu, H. Ma, X. Han, and B. Zhao, Mater. Horiz. 8 (2021). https://doi. org/10.1039/D0MH01356K.
(7) Y. Ozaki, Appl. Spectrosc. Rev. 24, 259–312 (1988).
(8) S. Andre, L.S. Cristau, S. Gaillard, O. Devos, E. Calvosa, and L. Duponchel, Anal. Chim. Acta. 892, 148–152 (2015).
(9) K.C. Doty and I.K. Lednev, TRAC-Trends Anal Chem. 103, 215–222 (2018).
(10) W.P. Findlay and D.E. Bugay, J. Pharm. Biomed. Anal. 16, 921–930 (1998).
(11) US Food and Drug Administration, PAT: A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance (FDA, Rockville, MD, 2004).
(12) A. Zarow, Dissertations. 270 (2011). https://digitalcommons.njit.edu/dissertations/270 (accessed on May 22, 2021).
(13) J.N. Sangshetti, M. Deshpande, Z. Zaheer, D.B. Shinde, and R. Arote, Arab. J. Chem. 10, S3412–S3425 (2017).
(14) B. Lenain, H. Lucas, C. Uerpmann, K.L. Davis, M.A. Ehly, M.S. Kemper, and I.R. Lewis, A whitepaper, Kaiser Optical Systems, 2021.
(15) F. Wang, J.A. Wachter, F.J. Antosz, and K.A Berglund, Org. Proc. Res. Dev. 4, 391–395 (2000).
(16) Y. Hu, J.K. Liang, A.S. Myerson, and L.S. Taylor, Ind. Eng. Chem. Res. 44, 1233–1240 (2005).
(17) D.S. Hausman, R.T. Cambron, and A. Sakr, Int. J. Pharm. 298, 80–90 (2005).
(18) D. Riolo, A Piazza, C. Cottini, M. Serafini, E. Lutero, E. Cuoghi, L. Gasparini, et al, J. Pharm. Biomed. Anal. 149, 329–334 (2018).
(19) K.A. Esmonde-White, M. Cuellar, C. Uerpmann, B. Lenain, and I.R. Lewis, Anal. Bioanal. Chem. 409, 637–649 (2017).
(20) B. Li, B.H. Ray, K.J. Leister, and A.G. Ryder, Anal. Chim. Acta. 796, 84–91 (2013).
(21) S. Li, Am. Pharm. Rev. (2010). https://www.americanpharmaceuticalreview.com/Featured-Articles/117400-Application-of-Online-Reaction-Monitoring-by-Raman-and-Infrared- Spectroscopy-in-Early-Drug-Development-Halogen-Lithium-Exchange- Chemistry/ (accessed on May 22, 2021).
(22) G.M. Hamminga, G. Mul, and J.A. Moulijn, Appl. Spectrosc. 61, 470–478 (2007).
(23) N.N. Misra, C. Sullivan, and P.J. Cullen, Curr. Biochem. Eng. 2, 4–16 (2015).
(24) M.B. Romia and M.A. Bernardez, in Spectroscopic Tools and Implementation Strategies for the Chemical and Pharmaceutical Industries, K.A. Bakeev, ed. (John Wiley & Sons Ltd., Hoboken, NJ, 2010), pp. 463–491.
(25) US Food and Drug Administration, Guidance for Industry: Development and Submission of Near Infrared Analytical Procedures Guidance for Industry (FDA, Rockville, MD, 2015).
(26) C. Pasquini, and S.H.F. Scafi, Anal. Chem. 75, 2270–2275 (2003).
(27) A.G. Dodda, K. Saranteas, and M.A. Henson, Org. Proc. Res. Dev. 19, 122–131 (2015).
(28) G.J. Gervasio, J. Chen, N.J. Falco, S.J. Sisk, and A. Slapikas, “Using Mass Spectrometry To Monitor Reactor Solvent Composition During Solvent Swaps” paper presented at the ISA 55th Analysis Division Symposium, New Orleans, LA, 2010.
(29) M. Ferrari, L. Mottola, and V. Quaresima, Can. J. Appl. Physiol. 29, 463–487 (2004).
(30) S. Doherty, and A. Garrett, Eur. Pharm. Rev. 4 (2005). https://www.europeanpharmaceuticalreview.com/article/2918/mass-spectrometry-pat
(31) J. Wasylyk, M. Huang, B. Wethman, and K. O’Connor, Pharm. Technol. 43, 35–39 (2019).
(32) K.R. Beebe, R.J. Pell, and M.B. Seasholtz, Chemometrics: A Practical Guide (John Wiley & Sons, New York, NY, 1998).
(33) “HoloReact Software Operations Manual, 2006852 R4” Kaiser Optical Systems.
(34) A. Belafriekh, F. Secundo, S. Serra, and Z. Djeghaba, Tetrahedron: Asymmetry 28, 473–478 (2017).
(35) M. Heshmat, A. Kazaryan, and E.J. Baerends, Phys. Chem. Chem. Phys. 16, 7315–7323 (2014).
Ravikumar Ramachandran and Karthik Jayaraman are with the Analytical Research & Development department at Biocon Bristol Myers Squibb Research and Development Center (BBRC), in Bangalore, India. Mallikarjun Narayanam is with Chemical Process Development–Analytical at Bristol Myers Squibb, in Moreton, UK. Robert Wethman is with Chemical Process Development at Bristol Myers Squibb, in New Brunswick, New Jersey. Amol G. Dikundwar is with the Department of Pharmaceutical Analysis at the National Institute of Pharmaceutical Education and Research (NIPER), in Hyderabad, India. Hemant Bhutani is with the Analytical R&D, Global Drug Development at Novartis Healthcare Private Limited, in Hyderabad, India. Direct correspondence to: karthik.j@syngeneintl.com or amol.dikundwar@niperhyd.ac.in.●

 Prove? A Look at Inorganic Analyses, Proficiency Tests, and Contamination and Error, Part I")










AI-Powered SERS Spectroscopy Breakthrough Boosts Safety of Medicinal Food Products
April 16th 2025A new deep learning-enhanced spectroscopic platform—SERSome—developed by researchers in China and Finland, identifies medicinal and edible homologs (MEHs) with 98% accuracy. This innovation could revolutionize safety and quality control in the growing MEH market.


New Raman Spectroscopy Method Enhances Real-Time Monitoring Across Fermentation Processes
April 15th 2025Researchers at Delft University of Technology have developed a novel method using single compound spectra to enhance the transferability and accuracy of Raman spectroscopy models for real-time fermentation monitoring.

Nanometer-Scale Studies Using Tip Enhanced Raman Spectroscopy
February 8th 2013Volker Deckert, the winner of the 2013 Charles Mann Award, is advancing the use of tip enhanced Raman spectroscopy (TERS) to push the lateral resolution of vibrational spectroscopy well below the Abbe limit, to achieve single-molecule sensitivity. Because the tip can be moved with sub-nanometer precision, structural information with unmatched spatial resolution can be achieved without the need of specific labels.



