Raman Spectroscopy as a Tool for Investigating Lipid-Protein Interactions
The authors discuss the feasibility of using Raman spectroscopic markers for the study of lipid-protein complexes in model systems and review relevant Raman spectroscopic markers for lipids and proteins.


A particular area of biophysical research in which Raman spectroscopy has great potential is the study of lipid–protein interactions. It is becoming increasingly clear that lipids can modulate membrane-embedded protein function and vice versa. This modulation can take both specific and nonspecific forms, but the underlying framework is defined by the amphiphatic nature of lipids and proteins. Proteins are embedded in the lipid bilayer by virtue of their hydrophilic and hydrophobic moieties expressed in the notion of hydrophobic coupling. Of special interest is the preferential enrichment of aromatic residues (for example, tryptophan [Trp]) in the interface between the hydrophilic aqueous phases and the hydrophobic bilayer core. The reason for this and its relation to hydrophobic coupling is not clear despite intensive research.
In this article, we will discuss the feasibility of using Raman spectroscopic markers for the study of lipid–protein complexes in model systems. Specifically, we will briefly review relevant Raman spectroscopic markers for lipids and proteins and how both kinds of markers can be used in the investigation of membrane-embedded proteins in fully hydrated liposomes. We will present data from a model system consisting of gramicidin incorporated into liposomes of various lipid compositions and discuss the feasibility of using spectroscopic markers for lipid and Trp residue markers.
The amphiphatic character of membrane-spanning proteins and lipid molecules implies that lipids and proteins self-assemble to avoid the high energetic cost of exposing hydrophobic moieties to water (1,2). The interaction — hydrophobic coupling — between lipids and membrane proteins is generally only slightly stronger than the interactions between lipids (3,4), suggesting an important role for unspecific lipid–protein hydrophobic interactions (3). The number of available high-resolution structures of integral membrane spanning proteins is increasing, and there is mounting evidence for regulation of membrane proteins by the host bilayer membrane through the hydrophobic coupling between membrane and proteins (4,5). This not only raises specific questions as to how membrane-spanning proteins interact with the lipid bilayer, but also opens the possibility to design biomimetic membranes based upon integral membrane proteins incorporated into encapsulated–supported systems for sensor and separation purposes (6).
One of the specific questions relates to a peculiar feature of integral membrane proteins: namely, a preference for aromatic (for example, Trp) residues located in the interfacial region between the hydrophobic core and the hydrophilic surface of the membrane. This has led to the notion of anchoring residues, in which the anchoring effect is thought to arise from the amphiphatic nature of interfacial aromatic residues (7,8). Despite considerable experimental progress using nuclear magnetic resonance (NMR), electron spin resonance (ESR), and molecular modeling (9–12) in the investigation of the effect of anchoring aromatic (and polar) residues, many issues remain unresolved.
Raman spectroscopy has immediate appeal in the study of lipid–protein interactions because it is a noninvasive and nondestructive technique, and the excitation wavelength can be chosen to be below the absorption frequency of water (13). However, Raman spectroscopy of biomembranes in aqueous solution is a technical challenge. The inherent weakness of Raman scattering and the low concentration of solvated samples makes the collection of a spectrum with a high signal-to-noise ratio difficult. But with increased sensitivity and recent developments in instrumentation, Raman spectroscopy is experiencing a revival in many research areas, including the study of lipids and lipid–protein complexes (14–19).
Here, we first discuss the technical challenges and relevant Raman spectroscopic markers for investigating the role of lipid–protein interactions in fully hydrated protein-vesicle samples. In particular, we present markers relevant for lipid molecular conformations and markers for the conformation of anchoring aromatic residues. Then we present a model system and give a few examples of how Raman markers appear in spectra of this system.
Raman Spectroscopy on Lipid–Protein Complexes in Aqueous Solution
Liposomes present a versatile tool for investigating lipid–protein interactions where one can fully control the lipid composition, protein content, and aqueous phases on both sides of the liposomal membrane. However, the use of protein-incorporated liposomes in Raman spectroscopy presents challenges compared to Raman spectroscopy on solid (powder) samples (19). Longer integration time is needed for aqueous solution samples and the intensity peaks appear broader (20). Also, containers used to avoid evaporation and contamination cause problems like loss of intensity and interferences, so windowless, liquid captive cells (21) are sometimes preferred (20). Raman spectroscopy on biochemical compounds isolated from living systems (and in particular, aromatic residues in membrane proteins) is made difficult due to autofluorescence, which can make the Raman bands "barely detectable" (20).
A longer excitation wavelength can alleviate the problem because fluorescence is frequency dependent. Excitation in the near-infrared (NIR) spectrum provides maximum intensity for a fluorescence-free spectrum and is therefore usually recommended for the study of biological samples. Comparing Raman excitations between 406 and 830 nm on tissues, the best defined lipid features are obtained at 782 and 830 nm (that is, with an excitation in the very near IR) (22). In fact, 785 nm seems to be the best suited excitation wavelength for the identification of a wide range of biomarkers (23). The excitation wavelength imposes constraints, such as the power available, the spot size, the exposure time, grating, and charge-coupled device (CCD) used. The comparison of mapping at 532 nm versus 785 nm illustrates the difference between actual collections at those two frequencies and the actual superiority of excitation in the NIR for this type of application (24).
Spectroscopic Markers for Lipids
The spectrum of lipids is to a large extent due to vibrations in the hydrocarbon chains, but vibrations in the head group region also can be used as specific lipid fingerprints. Here we will briefly present the acyl chain lipid markers (for example, in the CH deformation and the skeletal optical regions) and lipid head group markers.
Acyl Chain Markers
The CH deformation region includes the prominent CH bending mode at 1440 cm–1 for CH2 and 1460 cm–1 for CH3 (19,22). It is highly sensitive to lipid chain architecture and chains lateral packing (25). It has been used to detect the main phase transition of lipid vesicles (26). The other deformation prominent in the spectra is the CH twist. The CH2 twisting at 1296 cm–1 is found to decrease in intensity with increased CC stretching (22). The two other deformations of methylene groups, the wagging (out of plane) and the rocking (in plane), have not been assigned. For unsaturated chain segments, the =C-H in-plane deformation is at 1229 cm–1 and the =C-H out-of-plane deformation is at 970 cm–1 (22).
Another region of the spectrum marked by vibrations in the hydrocarbon chains is the CH stretch region located between 2800 and 3100 cm–1. The CH bond is highly localized and should therefore be rather insensitive to chain configuration or environment (25). However, the exact frequency seems to depend upon the methylene position on the chain (27). This region also has been shown to be sensitive to chain packing (28), probably due to Fermi resonance between the CH stretching and binary combination modes involving CH bending. The result is a broad feature that is difficult to interpret due to "a virtual continuum of binary combination states" (29). Despite the complexity of this region, assignments have been made. The peak at 2860 cm–1 is assigned to crystal mode interactions between adjacent hydrocarbon chains (30). The CH2 symmetric and antisymmetric stretching bands are located at 2840 cm–1 and 2880 cm–1, respectively (19). The symmetric CH2 stretching can be used as an indicator of the number of gauche chain conformers (60). The symmetric and asymmetric CH3 stretching bands are located slightly higher, around 2930 cm–1 and 2960 cm–1, respectively (19,22,25). The CH3 symmetric stretching is modified considerably with polarity (25) but is insensitive to perturbations of the terminal methyl group (30). For unsaturated chain segments, the =C-H stretch is located at 3010 cm–1 (19). The decrease of intensity ratio between the antisymmetric and the symmetric stretching modes associated with formation of gauche rotamers or the disruption of lateral packing interactions between hydrocarbon chains can be used to detect the main phase transition temperature (31). The CH region as a whole was used to detect subgel to gel transition (26), providing evidence that it can be used to investigate lipid chain packing.
The skeletal optical region of 1000–1200 cm–1 is highly sensitive to the hydrocarbon chain state (32). The CC stretching bands around 1133 cm–1 and 1064 cm–1 referred to as "all-trans skeletal vibrations" are associated with the all-trans chain segments (1119 cm–1 and 1079 cm–1, respectively according to Frank and colleagues [22]). A model for the contribution of trans chain segment sequences to the intensity at 1130 cm–1 is described in reference 33 and is thus linked to changes in acyl chain order-disorder (see also reference 28). The mode at 1080 cm–1 is probably a mixture of a C-C stretching and methylene wagging (34). The "gauche band" or "gauche marker" at 1089 cm–1 is associated with gauche chain segments. It is superimposed with the O-P-O symmetric stretch. The gauche band is found to shift and broaden during the main phase transition (25). For unsaturated chain segments, the C=C stretch is around 1660 cm–1 (1654 cm–1 for cis and 1669 cm–1 for trans) (22) and overlap with protein amide I mode (35).
Head Group Markers
Lower wavenumber bands than the regions discussed earlier also can be used for identification of the phospholipids. The band at 860 cm–1 has been assigned to the phosphate group common to all phospholipids, and a band at 1096 cm–1 has been assigned to the phosphodioxy group PO2– thought to depend upon the formation of H-bonds (19). The C=O stretch mode of the ester group of acyl chains depends upon environment: it is at 1739 cm–1 for acetic acid but 1729 cm–1 and 1744 cm–1 for two-acyl chain phospholipids (30). Also, the characterizing group has distinct markers: phosphatidylcholine lipids are identified by stretch vibrations in the choline group N+(CH3)3, and the stretching band at 717 cm–1 can be used as reference intensity, as can the antisymmetric stretch band at 875 cm–1 (19). Phosphatidylethanolamine lipids are identified by a minor band at 759 cm–1 associated to the ethanolamine group (19). Both the C-N symmetric stretching band and the PO2– band at 1096 cm–1 might have their intensity affected by cations (such as Ca2+ and Mg2+) linked to the ordering effect of these ions on lipid headgroups (25).
Other Lipid Regions
In Raman spectra of vesicle suspensions, a subtractable background appears. Apart from slow fluctuations in background that can look like apparent Raman bands (25) and the fluorescence due to impurities in a glass container, the main contribution to the background is water, despite the fact that water is mostly a weak Raman scatterer in the region investigated. The intense OH symmetric stretching of water at 3400 cm–1 and asymmetric stretching at 3455 cm–1 appears as a shoulder in the CH stretch region. The bending at 1640 cm–1 varies, depending upon the extent of hydrogen bonding (36,37). Apart from the intramolecular bands, there are intermolecular libration bands from water (38), which form a broad envelope from 300 to 1000 cm–1 (37,39) including rocking at 440 cm–1, twisting at 550 cm–1 or 650 cm–1, and wagging at 730 cm–1. All three are found to decrease in wavenumber with increasing temperature (37). In addition, there are weak unassigned bands between 1000 cm–1 and 2600 cm–1 at 1300 cm–1, 1555 cm–1, 2118 cm–1, and 2390 cm–1 (36,39).
Protein Spectroscopic Markers
The bands can be divided into two groups: the main-chain bands (amide bands) related to the protein backbone and the bands related to the side chains, where we will focus on the Trp side chains.
The amide I and amide III bands are classical markers for the conformation of proteins (40). The α-helix, β-sheet, and random coil secondary structural motifs have different peak wavenumbers and band shapes, and the composition of the protein in terms of these secondary structures can be calculated from these bands (41,42). However, using the amide I band for structural analysis might be difficult, necessitating a combination of experiments with classical and quantum methods for advanced interpretation. The amide I mode usually is described as CO stretching mode with some mixture of CCN deformation (43). The exact wavenumber decreases with hydrogen bonding to the carbonyl group. The amide III is a CN stretch and NH in plane bending mode with C(C)H3 symmetric bending and CO in plane bending contributions between 1200 and 1300 cm–1. The exact wavenumbers depend upon environment and structure, and for example, the peak at 1263 cm–1 depends upon the dihedral angle ψ (44). Amide II, although less structure sensitive than amide I and III, could be used in theory for detecting protein conformation, but in practice it is too weak.
A nonamide mode at 1395 cm–1 referred to as the amide S mode has been shown to arise from Cα(C)H bending with contribution from CN stretch and NH in plane bending. It is enhanced by coupling to the amide III mode via CH bending and CC stretching. When the Cα-H bond is rotated, there is uncoupling of the two modes and disappearance of the amide S band. The Cα-H bond is oriented cis to the carbonyl group in α-helices and trans in loop or β-sheet structures. The intensity of the amide S band can thereby be linearly related to the nonhelical content of the protein. It has been used for the structure analysis of proteins (42).
Raman Markers for Trp
Apart from the main chain bands, protein spectra also have bands specific to the amino acid side chains (for example, NH stretch at 3300 cm–1, CH stretch at 2800–3000 cm–1, and CH bending at 1450 cm–1). Aromatic groups are rich in electrons and therefore prominent in spectra, and Trp has several bands that have been reported to be suitable as conformational markers (45). The assignments have been confirmed by computer simulation (46).
The peak around 1542 cm–1 can be used as a conformation marker for the orientation of the Trp residue because the exact wavenumber (υW3) of the peak is determined by the angle χ2,1 (47):


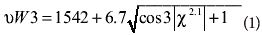
The intensity ratio of the doublet at 1340 cm–1 and 1360 cm–1 I1360/I1340 is a marker (denoted W7) of the Trp's environment hydrophobicity: the stronger the hydrophobicity, the higher the ratio. The participation of Trp in H-bonding as a hydrogen donor can be monitored using the markers at 877 cm–1 (denoted W17), 1490 cm–1 (denoted W4), and 1430 cm–1 (denoted W6) (45,47). For W17, a scale has been proposed in which 883 cm–1 corresponds to a non-H-bonded state, 877 cm–1 to a medium strength H-bonding, and 871 cm–1 to strong H-bonding (47).
Model System
Spectra of living systems and macromolecules are highly complex superimpositions of bands differing only slightly from one another as they originate from identical chemical groups. Often, overlapping bands make the interpretation of spectra difficult. A model system such as lipid vesicles with incorporated model proteins and peptides has fewer components than a living cell, making the interpretation of Raman spectra feasible.


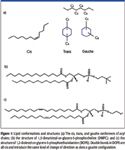
Figure 1
A well-suited model system in which to investigate the role of interfacial aromatic residues for the hydrophobic lipid–protein interactions is gramicidin incorporated into lipid bilayers. In lipid environments, the 15-amino-acid gramicidin sequence adopts a β6.3–helical fold with four interfacial Trp residues (48) (Figure 2). Two monomers can join by hydrogen bonding and form an antiparallel dimer. This arrangement results in a transmembrane protein with a hydrophobic length l of about 22 Å spanning the bilayer (49,50). High-resolution structures of gramicidin in the β6.3–helical structure favored in lipid environments are available (51), and Raman bands for gramicidin in the crystalline state have been assigned (52,53).
Whenever gramicidin dimers are incorporated in bilayers having a hydrophobic thickness d0 larger than the hydrophobic length of the gramicidin dimer channel, there will be a hydrophobic mismatch, and this mismatch leads to local protein-induced bilayer deformation (that is, lipid monolayer bending and compression) (3,54,55) (Figure 2).


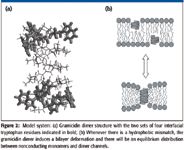
Figure 2
Changes in acyl chain conformation, whether induced by monomers or dimers, should in principle be visible as changes in the lipid Raman markers discussed earlier and exemplified in the relative porportion of gauche versus trans chain segments. The Trp "interfacial anchoring" effect seems to be related to lipid species because molecular dynamics (MD) simulations suggest that the Trp residues of gramicidin can interact preferentially with the phosphatidylethanolamine (PE) lipid headgroups rather than with phosphatidylcholine lipid headgroups (10). Thus, Raman markers for Trp rotameric states and H-bonding will in principle provide information about how this anchoring is coupled to lipid head group specifcity and hydrophobic mismatch.
Materials and Methods
The saturated dimyristoylphosphatidyl choline (DMPC) and the unsaturated dioleoylphosphatidy-ethanolamine (DOPE) (Avanti Lipids, Alabaster, Alabama). are both in the liquid state at room temperature. DMPC and DOPE:DMPC (3:1 mol:mol) vesicles were prepared using standard protocols (56) to a final aqueous concentration of 10 mg/mL. Gramicidin (Sigma Aldrich, St. Louis, Missouri) in powder form was dissolved in ethanol. The final gramicidin:lipid ratio in DMPC and DOPE:DMPC (3:1) vesicles was 4:100 (mol:mol).
The measurements were made using an inVia Raman microscope with 50× objective (Renishaw plc, Old Town, U.K.) with a laser wavelength of 785 nm. A maximal effect of 300 mW and a grating with 1200 lines/mm was used. A quantity of 200 μL of lipid suspension was placed on a glass cover slip in the microscope stage. The microscope objective was lowered until it came in contact with the drop and subsequently raised several mm without breaking contact with the drop. An extended mode scan of 1–5 min was used at maximum power. To improve the signal-to-noise ratio, up to 10 scans were accumulated, depending on the sample. The spectra were normalized to the band at 717 cm–1. The background was approximated by fast Fourier averaging of the spectra where the prominent bands had been removed and subtracted from the spectra. For the inserts in Figures 3b and 3d, the spectra were smoothed using adjacent averaging. Subtraction, Gaussian peak fitting, and smoothing routines were performed using Origin Pro 8 analysis software (OriginLab, Northampton, Massachusetts).



Figure 3
Results
We investigated the gramicidin-induced changes in acyl chain conformation through the bands sensitive to lipid chain conformations. Then we analyzed the role of lipid headgroups (bilayer composition) on the change in conformation of the bilayer lipids. Finally, we illustrated how the Trp bands for gramicidin are affected by lipid bilayer composition.
Gramicidin Induces Gauche Conformational Acyl Chain States
The perturbation of bilayer lipids on gramicidin binding was investigated for a DMPC bilayer and a DOPE:DMPC (3:1) bilayer by comparison of the Raman intensity of the bands dependent upon lipid chains conformation and packing in the absence and in the presence of gramicidin. The Raman spectra of DMPC vesicle suspensions in the absence and presence of gramicidin are given in Figures 3a and 3b, respectively. The Raman spectra of DOPE:DMPC (3:1) mixture vesicles suspensions in the absence and presence of gramicidin are given in Figures 3c and 3d, respectively.
All of the regions sensitive to lipid chains conformation are modified upon binding of gramicidin in both bilayers. In the CH bend region, the intensity at 1440 cm–1 is more than halved in the DMPC–gramicidin bilayer compared to DMPC alone. In the CC stretch region, the intensity at 1130 cm–1 related to the number of trans segments in the chains is almost halved upon gramicdin incorporation. The ratio I1089/I1128 quantifying the number of gauche bonds (relative to trans bonds) increases by 40% in DMPC with gramicidin. In the CH2 twist region, the intensity at 1296 cm–1, which decreases with the number of trans chain segments, is decreased fourfold in DMPC upon gramicidin incorporation. In the CH stretch region, the amplitude of the peak at 2850 cm–1 is increased by 30% in DMPC when gramicidin is present.
The quality of the spectra in DOPE:DMPC (3:1) alone and with gramicidin incorporated are not as good as the DMPC and DMPC–gramicidin spectra. The skeletal region is difficult to analyze, and it is therefore not possible to quantify any changes in gauche acyl chain conformations. However, the CH stretch at 2850 cm–1 is reduced about 15% in DOPE:DMPC (3:1) upon gramicdin incorporation, in contrast to the observation for DMPC–gramicidin versus DMPC.
Tryptophan Rotameric States and H-Bonding Depend on Lipid Composition
The conformational state of Trp residues of gramicidin as a function of the host lipid bilayer was investigated by comparing the Raman Trp markers in the spectra shown in Figure 3.
The W3 conformational marker is centered at 1554 cm–1 in DMPC–gramicidin, corresponding to a |χ 2,1| of around 100° (Figure 3b). It has a bimodal distribution in DOPE:DMPC (3:1)–gramicidin, with approximately two-thirds of the intensity around 1558 cm–1 corresponding to a |χ 2,1| of around 120°, and one-third of the intensity around 1545 cm–1 corresponding to a |χ 2,1| of around 80° (Figure 3d).
For W17 in DMPC–gramicidin, Gaussian fitting reveals a trimodal distribution with most of the intensity centered on 876 cm–1 and two lesser peaks at 882 cm–1 and 889 cm–1, indicating that on average, most tryptophan residues engage in medium strength H-bonding, whereas a smaller population tend to be non-H-bonded (the band at 883 cm–1) (see insert in Figure 3b.) In DOPE:DMPC (3:1)–gramicidin, the spectral region also has a trimodal distribution, with most of the intensity centered on 876 cm–1 and two lesser peaks at 864 cm–1 and 886 cm–1. This corresponds to a medium-to-strong H-bonding (stronger and broader band at 877 cm–1) but with a possible contribution from nonbonded interactions (see insert in Figure 3d).
The W7 environmental hydrophobicity marker I1360/I1340 is difficult to anlyze in the spectra because there are additional lipid bands in that region (Figures 3a and 3c). DMPC I1360/I1340 appears to be about unity, indicating low hydrophobicity. In DOPE:DMPC (3:1)–gramicidin I1360/I1340 may be <1, but the quality of the spectra is not sufficiently good to conclude further on this marker.
Discussion
Lipid Conformation
There is an increase in the number of gauche conformations when gramicidin is incorporated in DMPC. The increase in the trans-to-gauche ratio has the same magnitude as the disordering observed during the main phase transition or during a large increase in temperature in the fluid phase (57). An increase in lipid chain disorder likely reflects a compression of the lipids associated with gramicidin channels to compensate for the hydrophobic mismatch. Because each gramicidin monomer will have about 10 annular lipids (that is, 20 for a gramicidin dimer channel), a gramicidin:lipid ratio of 4:100 implies that 40% of all the lipids are in direct contact with a monomer and that only a few (<3) lipids are "traversed" when going from one gramicidin monomer–dimer to the other. Thus, a considerable portion of lipids can be compressed but at the same time motionally restricted. This is consistent with observations using spin-labeled lipids, where addition of gramicidin results in a decreased motionally averaging (58).
Trp Conformation and Environment
The W3 marker seems most promising in lipid–protein analysis here, showing re-orientation of the Trp residues, whereas the W17 and W7 markers are more difficult to use due to lipid bands in these spectral regions. While W7 cannot be anlyzed using the spectra obtained, W17 suggests that there is a small increase in H-bonding when going from a DMPC bilayer to a DOPE:DMPC (3:1) bilayer. A reason for this could be that the DOPE lipids can engage in carbonyl-Trp H-bonding (and more cation-π interactions [10]) with the Trp indole as the PE moity can be closer to the indole than the PC moiety. The closer packing will thus tend to make tryptophan residues more accessible to lipid H-bonding but less accessible to water H-bonding. This is also consistent with the observation that gramicidin Trp becomes less accessible to water and is deuterated more slowly when going from DMPC to DPPC (that is, with an increase in hydrophobic mismatch [45]). Although the choline group stretch vibration at 875 cm–1 precludes an exact H-bonding estimate based upon W17, we note that the lower molar content of choline in DOPE–DMPC in its own right would dimish the 875 cm–1 band, but we observe an increase in that region when going from DMPC to DOPE:DMPC (3:1), suggesting that H-bonding is increased. The conformational changes also could be due to gramicidin tilt adaptation to mismatch (59) because this could cause reorientation of the Trp and change their H-bonding capability. However, the orientation of gramicidin Trp residues does not change when comparing DLPC with DMPC (that is, when the hydrophobic mismatch is decreased [45]).
Outlook
We have used a relatively simple way to measure Raman scattering on vesicles: measuring during prolonged times on concentrated suspensions. An improvement leading to better spectra would be to use optical trapping on a single large vesicle using a Raman microscope (15). Another way to improve the approach would be to combine Raman spectroscopy with "Raman-complementary" IR spectroscopy. In IR, it should be possible to identify the well-defined wagging bands that correspond to different conformations (gauche end, kink, double gauche) (34). The methylene and methyl stretching modes can confirm the change in the relative number of trans and gauche conformations on peptide binding obtained by Raman spectroscopy (60). Future work also should include a temperature study to determine what the intensity is in the all-trans configuration (57). Finally, IR phosphate bands can be related to H-bonding and thereby report on headgroup conformation (25,61).
Raman spectroscopy of vesicle suspensions shows that gramicidin affects and is affected by the bilayer lipids. Gramicidin incorporation increases the number of gauche bonds in the lipid acyl chains, indicating an increase in acyl chain compression. Raman spectroscopic markers for the Trp residues of gramicidin (in particular W3) show that Trp rotametric states (and perhaps H-bonding) are influenced by the lipid composition of the host bilayer.
Acknowledgments
The authors would like to thank Erik Skibsted at Novo Nordisk A/S for providing access to their Raman equipment. This work was supported by a grant from the Danish National Research Foundation to QUP. Claus Hélix Nielsen also received additional support through MEMBAQ, a Specific Targeted Research Project (STREP), by the European Commission under the Sixth Framework Programme (NMP4-CT-2006-033234) and from WATERMEMBRANE, a project supported by The Danish National Advanced Technology Foundation (023-2007-1).
Frederic N.R. Petersen is with the Quantum Protein Center, Department of Physics, Technical University of Denmark, Lyngby, Denmark. Claus Hélix Nielsen is with the Quantum Protein Center, Department of Physics, Technical University of Denmark, Lyngby, Denmark, and Aquaporin, A/S, Lyngby, Denmark.
References
(1) K.A. Sharp, A. Nicholls, R.F. Fine, and B. Honig, Science 252, 106–109 (1991).
(2) J.N. Israelachvili, S. Marcelja, and R.G. Horn, Q. Rev. Biophys. 13, 121–200 (1980).
(3) C. Nielsen, M. Goulian, and O.S. Andersen, Biophys. J. 74, 1966–1983 (1998).
(4) J.A. Lundbaek, J. Phys.: Condens. Matter 18, S1305–S1344 (2006).
(5) C.H. Nielsen, Lipid-protein interactions in biomembranes. In: H.G. Bohr, Ed., Handbook of Biophysics (Wiley, Berlin, 2009), pp. 329–358.
(6) J.S. Hansen, M.E. Perry, J. Vogel, T. Vissing, O. Geschke, J. Emneus, and C.H. Nielsen, J. Micromech. Microeng. 19, 025014 (2009).
(7) M. Schiffer, C.-H. Chang, and F.J. Stevens, Protein Eng. 5, 213–214 (1992).
(8) J.A. Killian and T.K. Nyholm, Curr. Opin. Struct. Biol. 16, 473–479 (2009).
(9) E. Strandberg, S. Ozdirekcan, D.T. Rijkers, P.C. van der Wel, R.E. Koeppe, 2nd, R.M. Liskamp, and J.A. Killian, Biophys. J. 86, 3709–3721 (2004).
(10) F.N. Petersen, M.O. Jensen, and C.H. Nielsen, Biophys. J. 89, 3985–3996 (2005).
(11) D. Marsh and L.I. Horvath, Biochim. Biophys. Acta 1376, 267–296 (1998).
(12) M.O. Jensen, O.G. Mouritsen, and G.H. Peters, Biophys. J. 86, 3556–3575 (2004).
(13) R. Petry, M. Schmitt, and J. Popp, ChemPhysChem. 4, 14–30 (2003).
(14) R.J. Meier, Chem. Soc. Rev. 34, 743–752 (2005).
(15) J.M. Sanderson and A.D. Ward, Chem. Commun. (Camb.) 1120–1121 (2004).
(16) C. Lee and C.D. Bain, Biochim. Biophys. Acta 1711, 59–71 (2005).
(17) C.H. Nielsen, S. Abdali, J.A. Lundbæk, and F. Cornelius, Spectroscopy 22(2), 52–63 (2007).
(18) C. Krafft, Anal. Bioanal. Chem. 378, 60–62 (2004).
(19) C. Krafft, L. Neudert, T. Simat, and R. Salzer, Spectrochim Acta, Part A 61, 1529–1535 (2005).
(20) S.E.M. Colaianni, J. Aubard, S.H. Hansen, and O. Faurskov Nielsen, Vib. Spectrosc. 9, 111–120 (1995).
(21) M.H. Brooker, O. Faurskov Nielsen, and D. Christensen, J. Raman Spectrosc. 26, 331–334 (2005).
(22) C.J. Frank, D.C. Redd, T.S. Gansler, and R.L. McCreery, Anal. Chem. 66, 319-326 (1994).
(23) S.E. Villar, H.G. Edwards, and M.R. Worland, Orig. Life Evol. Biosph. 35, 489–506 (2005).
(24) J.R. Beattie, S. Brockbank, J.J. McGarvey, and W.J. Curry, Mol. Vis. 11, 825-832 (2005).
(25) D.F. Wallach, S.P. Verma, and J. Fookson, Biochim. Biophys. Acta 559, 153–208 (1979).
(26) C.B. Fox, G.A. Myers, and J.M. Harris, Appl. Spectrosc. 61, 465–469 (2007).
(27) I.R. Hill and L. I.W., J. Chem. Phys. 70, 842–851 (1979).
(28) B.J. Bulkin and N. Krishnamachari, J. Am. Chem. Soc. 94, 1109–1112 (1972).
(29) J.H. Schachtschneider and R.G. Snyder, Spectrochim. Acta 19, 117–168 (1963).
(30) B.P. Gaber, P. Yager, and W.L. Peticolas, Biophys. J. 24, 677–688 (1978).
(31) T. Taraschi and R. Mendelsohn, Proc. Natl. Acad. Sci. U.S.A. 77, 2362-2366 (1980).
(32) J.L. Lippert and W.L. Peticolas, Proc. Natl. Acad. Sci. U.S.A. 68, 1572–1576 (1971).
(33) D.A. Pink, T.J. Green, and D. Chapman, Biochemistry 19, 349–356 (1980).
(34) R.G. Snyder, J. Am. Chem. Soc. 47, 1316–1360 (1967).
(35) C. Krafft, S.B. Sobottka, G. Schackert, and R. Salzer, Analyst 130, 1070–1077 (2005).
(36) G.E. Walrafen and E. Beitz, J. Chem. Phys. 59, 2646–2650 (1973).
(37) D.M. Carey and G.M. Korenowski, J. Chem. Phys. 108, 2669–2675 (1998).
(38) G.E. Walrafen, J. Chem. Phys. 40, 3249–3256 (1964).
(39) M. Moskovits and K.H. Michaelian, J. Chem. Phys. 69, 2306–2311 (1978).
(40) G. Socrates, Biological molecules — Macromolecules. In: G. Socrates, (Ed.), Infrared and Raman Characteristic Group Frequencies (John Wiley & Sons, 2001), pp. 328–341.
(41) K. Griebenow and A.M. Klibanov, J. Am. Chem. Soc. 118, 11695–11700 (1996).
(42) R. Schweitzer-Stenner, Vib. Spectrosc. 42, 98–117 (2006).
(43) N.G. Mirkin and S. Krimm, J. Mol. Struct. 242, 143–160 (1991).
(44) T. Jordan and T.G. Spiro, J. Raman Spectrosc. 25, 537–543 (1994).
(45) H. Takeuchi, Y. Nemoto, and I. Harada, Biochemistry 29, 1572–1579 (1990).
(46) A. Combs, K. McCann, D. Autrey, J. Laane, S.A. Overman, and G.J. Thomas, J. Mol. Struct. 735–736, 271–278 (2005).
(47) H. Takeuchi, Biopolymers 72, 305–317 (2003).
(48) K. He, S.J. Ludtke, Y. Wu, H.W. Huang, O.S. Andersen, D. Greathouse, and R.E. Koeppe, II, Biophys. Chem. 49, 83–89 (1994).
(49) A.S. Arseniev, A.L. Lomize, I.L. Barsukov, and V.F. Bystrov, Biol. Membr. 3, 1077–1104 (1986).
(50) O.S. Andersen, C. Nielsen, A. Maer, J.A. Lundbaek, M. Goulian, and R.E. Koeppe II, Methods Enzymol. 294, 208–224 (1999).
(51) O.S. Andersen, Ann. Rev. Physiol. 46, 531–548 (1984).
(52) V.M. Naik and S. Krimm, Biophys. J. 49, 1147–1154 (1986).
(53) V.M. Naik and S. Krimm, Biochem. Biophys. Res. Commun. 125, 919–925 (1984).
(54) H.W. Huang, Biophys. J. 50, 1061–1070 (1986).
(55) P. Helfrich and E. Jakobsson, Biophys. J. 57, 1075–1084 (1990).
(56) F. Szoka, Jr., and D. Papahadjopoulos, Annu. Rev. Biophys. Bioeng. 9, 467–508 (1980).
(57) D. Marsh, Biochim. Biophys. Acta 363, 373–386 (1974).
(58) M. Ge and J.H. Freed, Biophys. J. 65, 2106–2123 (1993).
(59) J.A. Killian, Biochim. Biophys. Acta 1376, 401–415 (1998).
(60) C.J. Steer, J.S. Vincent, and I.W. Levin, J. Biol. Chem. 259, 8052–8055 (1984).
(61) F.M. Goni, and J.L. Arrondo, Faraday Discuss. Chem. Soc. 117–126 (1986).





















Smarter Sensors, Cleaner Earth Using AI and IoT for Pollution Monitoring
April 22nd 2025A global research team has detailed how smart sensors, artificial intelligence (AI), machine learning, and Internet of Things (IoT) technologies are transforming the detection and management of environmental pollutants. Their comprehensive review highlights how spectroscopy and sensor networks are now key tools in real-time pollution tracking.

