Understanding the Fundamental Components of Sample Introduction for ICP-OES and ICP-MS
Analytical techniques based on inductively coupled plasma (ICP) are commonly used for analyzing the elemental composition of a sample and are widely applied in analytical testing laboratories. Particularly for larger laboratories, analysis time and throughput are essential performance indicators, which can be severely impacted if sample analysis needs to be repeated because of insufficient stability or quality control (QC) failures. However, such failures are, in many cases, avoidable by selecting the right configuration of the sample introduction system of the instrument. This tutorial explains the most critical components of the sample introduction system of modern ICP-optical emission spectroscopy (OES) and ICP-mass spectrometry (MS) instruments, providing analysts with a guide for initial configuration settings and recommended maintenance intervals for reliable daily operation.
Inductively coupled plasma (ICP), when combined with optical emission spectroscopy (OES) and mass spectrometry (MS), is a well-established tool for elemental analysis. Continued technological development has led to significant increases in sensitivity and interference removal (for common polyatomic as well as other types of interferences). However, the technology surrounding sample introduction has remained comparable over the years.
Many of the challenges analysts face today, such as signal suppression, drift, or even failure of quality control (QC) checks can be related to the sample introduction system setup. In many cases, incorrect configuration or errors during routine maintenance are the root cause for these issues.
Unexpected Contamination: Sample Vials and Tubing
Techniques such as ICP-OES and ICP-MS provide the high sensitivity required to analyze the concentrations of toxic metals at trace and ultratrace levels. However, high sensitivity makes both techniques susceptible to problems associated with unintended contamination, such as increased background levels, elevated detection limits, or over and underestimated results. There are multiple ways in which a sample can become contaminated, including, but not limited to, sample preparation, workplace cleanliness, and the purity of reagents used (that is, acids used in digestion or dilution steps). However, in most cases, the impact of plasticware used in the laboratory is underestimated. Plasticware comprised of perfluoroalkoxy (PFA) polymers is known to be available in high purity, whereas vials that are used for sample storage are often made from polypropylene (PP) or polyethylene (PE), which have lower purity.
Before starting an analysis, it is good practice to look at the quality of all materials and reagents used in the sample preparation process. Contamination can arise from the surrounding laboratory environment (even in areas dedicated to trace metal analysis) where sporadic and unexpected contamination can occur. Usually, this contamination can be attributed to residues in the plasticware used, such as vessels or peristaltic pump tubing. Many laboratories refrain from switching suppliers of sample tubes because it can potentially introduce contamination because of differences in the manufacturing processes and materials. Although most tubes are fairly clean, experience has shown that vial caps can be a significant source for metal contamination, particularly aluminum, zinc, nickel, and copper. The extent of contamination can be significantly reduced by rinsing vials and caps with dilute nitric acid prior to use. The same considerations may apply to peristaltic pump tubing (typically made from polyvinyl chloride [PVC]). A brief cleaning or conditioning with an acidic solution (a few minutes is sufficient) can wash out any unwanted traces of metals before they can be introduced into the sample analysis.
Sample Consumption and Handling: Peristaltic Pump, Nebulizer, and Spray Chamber
Problems with an incorrect sample delivery to the plasma often express themselves in a secretive manner. An important parameter to be checked during the daily performance check is the short-term stability of the system, or percent relative standard deviation (%RSD), of the signal determined over a period of 10 min. Some laboratories or methods require continuous monitoring of the signal stability in each run, where data showing a %RSD greater than 2–3% must be discarded. In many cases, instabilities observed either during set up of the instrument or the sample analysis are related to the sample delivery to the plasma. The most common source of instability is related to the peristaltic pump delivery speed, the worn out peristaltic pump tubing, or the uneven draining of the spray chamber. When signal variation appears to be periodic (a similar pattern repeating every few seconds), sample delivery is most often found to be the source of the problem.
If increased signal fluctuation is only observed for a subset of analytes, the root cause can sometimes be found in the preparation of the sample. Elements, such as aluminum or iron, need to be stabilized in an acidic environment. If they are not, they may form colloid species like hydroxides, which can lead to unstable signals in both ICP-OES and ICP-MS. This problem can be observed with samples having a pH value of approximately 4, where additional acid is needed to lower the pH further to improve the stability of the signal.
When Problems Become Visible in ICP-OES: Torch
Have you ever noticed the central channel of your plasma turn orange when running an analysis? The orange glow is emission from excited sodium atoms and is a sign that your sample contains high levels of dissolved solids (typically dissolved salts of mainly sodium, potassium, magnesium, and calcium). In addition, the presence of organic solvents is immediately visible in an incorrectly configured system. In the latter case, the plasma exhibits a green emission followed by an orange glow from the carbon deposited at the sample cone tip, with the plasma often extinguishing after a short time. All instruments available on the market allow at least a visual observation of the plasma using a window. Certain instruments allow direct observation of the plasma directly on the control PC by means of a camera. In either case, a close look at the plasma when challenges are observed may help to identify issues with the analysis. In many cases, problems related to the analysis of difficult sample types, such as brine or organic solvents, can be overcome by selecting a different configuration of the sample introduction system configuration. Table I contains a few recommendations for analysis carried out using an ICP-OES instrument. ICP-MS is generally able to handle samples containing completely dissolved solids of up to 0.2% (m/v). Higher total dissolved solids can be handled by an ICP-MS instrument, but it would require a special configuration using argon gas from the instrument as a diluent that is introduced to the sample flow at the spray chamber. If the samples contain hydrofluoric acid (HF), then all components of the sample introduction system need to be exchanged for HF-resistant versions.


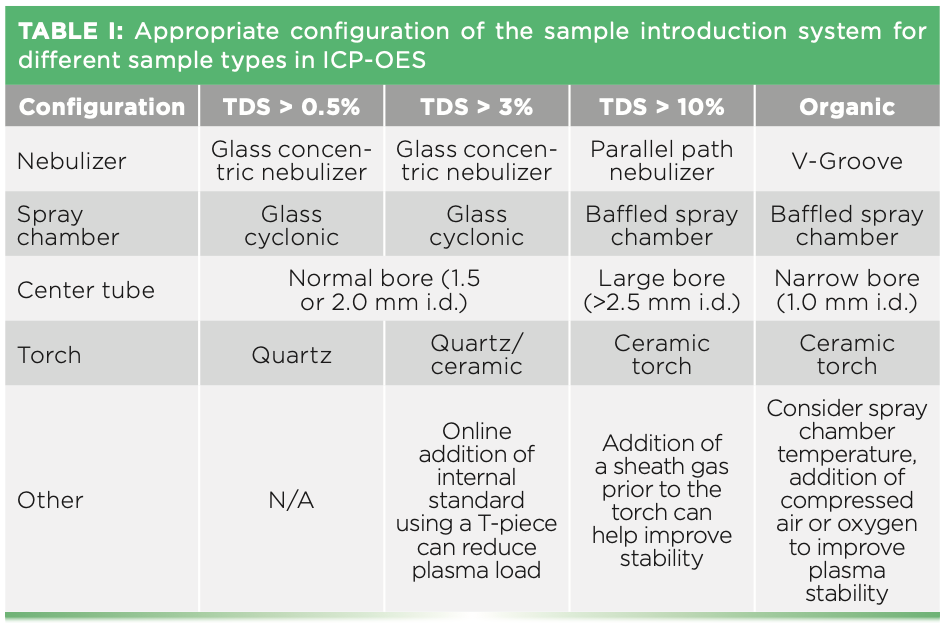
More Than Just an Interface: The Role of Cones in ICP-MS
Although ICP-OES and ICP-MS have a lot in common when looking at the general setup of the sample introduction system, they differ significantly when it comes to the most important sample types. ICP- OES is able to analyze high matrix-containing samples such as brines and wastewater. On the other hand, ICP-MS is preferably applied for sample types that contain less problematic matrices or those that have undergone thorough cleanup procedures to remove the matrix components. The main reason for this distinction is that in ICP-MS, ions need to be transferred into the high vacuum of the mass spectrometer, whereas the emission of light can be viewed at an ambient atmosphere (residual air needs to be removed inside the optical system though to avoid emission signal absorption, particularly for wavelengths in the UV range of the spectrum). The ion extraction is accomplished in the interface with a set of two narrow orifices (less than 1 mm diameter) kept at an intermediate pressure level of less than 2 mbar. The interface cones of an ICP-MS can typically handle up to 0.2% (m/v) dissolved solids entering the plasma. Above this level, blocking of the orifices at the interface will occur, particularly on the skimmer cone where its cooler temperature makes it more prone to clogging from the buildup of accumulated solids. As mentioned previously, sample analysis with a higher matrix load by ICP-MS is possible with the introduction of argon gas as a diluent between the spray chamber and the torch. Although no manual dilution with a liquid is made, the sample flow is reduced, thus reducing the amount of matrix introduced. Lower sample flow results in a reduction in sensitivity of the system, but the impact on the method detection limits (MDL) is often not noticeable compared to a manual or automated liquid dilution. The direct analysis of samples (without prior manual dilution) is attractive as less diluents are used, reducing cost, labor, and the risk of errors or contamination. However, this approach is less flexible, with the dilution factor remaining constant for all the samples in a batch and samples containing less challenging matrix being analyzed under the same conditions (that is, reduced sample flow) as the more challenging samples. In addition, automated adjustment of the dilution factor (based on the concentration found in a sample relative to the calibrated range) is not possible as this causes a mismatch between the calibration solution signal response and the diluted (with a different dilution factor, that is not exactly calculated under argon gas dilution conditions) sample signal response.
Problems at the interface region can often be recognized by checking the response of the internal standards. Internal standards are elements that are not present (or present at very low levels) in samples. They are added, at a fixed concentration, to all blanks, calibration solutions, and samples, either on-line via a mixing tee or valve port (easy), or off-line, by manual addition (tedious). When a sample contains high concentrations of sodium, potassium, and calcium, the internal standard signals are suppressed compared to what they are in the blank or matrix-free solutions. Many regulated methods specify how much internal standard suppression is permitted in a sample, which is typically between 75–80% of the signal observed in a sample matrix-free blank (usually the first calibration blank in the analysis). In general, if internal standard recovery is less than 75–80% in any sample, dilution and re-analysis is required. The most common reason for internal standard recoveries being greater than 120% is often attributed to the ICP-MS interface not being conditioned properly. Conditioning is the process of running a dummy sample containing similar matrix to the samples to be analyzed through the instrument for approximately 30 min before you start the actual analysis, which causes the interface cones to become coated and helps to stabilize ion signals and minimize drift. It remains somewhat unclear as to why cone conditioning helps improve signal stability, but it is a well-documented fact that it does and conditioning in this way is standard practice. Cone conditioning is especially important after cleaning the interface cones or when they are replaced. In general, cleaning the interface cones should only be considered when signs of blockage or unremovable contamination are observed or if the instrument performance has significantly dropped.
Finally, The Route to Success
Finding the right configuration of the sample introduction system for a specific sample type is easy once a few questions are answered up front. The most important questions are related to the sample matrix: is it aqueous or organic? What is the amount of total dissolved solids? And what is the approximate composition?
Figure 1 summarizes a few of the crucial aspects that should be considered when setting up an instrument (either ICP-OES or ICP-MS) for analyzing a new sample type.


FIGURE 1: The route to success in managing the analysis of previously unknown sample types.
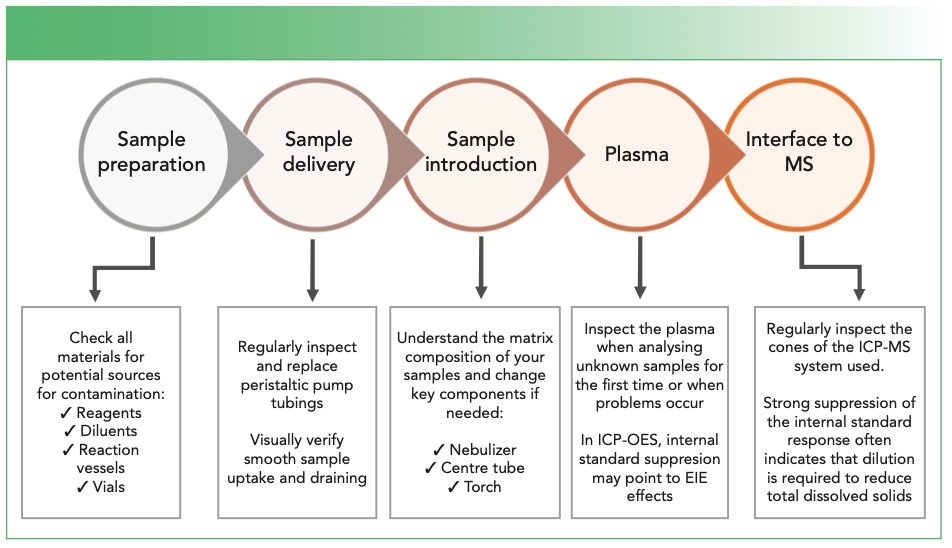
If a method is defined and validated but does not perform up to expectations, it is important to know which key areas of the system configuration to focus on and to understand how the different components of the sample introduction system are working together. As a general rule, instability is usually caused by problems upstream of the sample introduction system, whereas drift and suppression are rather caused downstream (injector tube, torch, and interface cones [in the case of ICP-MS]). In some cases, effects that may be attributed to the hardware may also originate from unexpected interferences from the sample, but that is a subject for another tutorial.
Simon Nelms and Daniel Kutscher are with Thermo Fisher Scientific. Direct correspondence to: daniel.kutscher@thermofisher.com ●




 Bonds")













Atomic Perspectives: Highlights from Recent Columns
March 3rd 2025“Atomic Perspectives,” provides tutorials and updates on new analytical atomic spectroscopy techniques in a broad range of applications, including environmental analysis, food and beverage analysis, and space exploration, to name a few. Here, we present a compilation of some of the most popular columns.


Pittcon 2025: Highlighting Talks on Atomic Spectroscopy
February 26th 2025At Pittcon this year, there will be numerous sessions dedicated to spotlighting the latest research that uses atomic spectroscopy or elemental analysis techniques. We highlight some of these talks below that might pique the interest of spectroscopists and researchers attending the conference this year.

