Fully Integrated Analysis of Metabolites, Impurities, and Degradants Using LC–NMR–MS
Combining the three techniques of LC, MS, and NMR into one integrated system provides optimal use of NMR intrument time by using information-rich MS data to automatically guide the NMR operation. Here, the authors explore just this type of system.



Many situations exist in which the unequivocal structural identity of an impurity, a degradant or a metabolite is required in the shortest possible time. Unknown impurities can stop multimillion dollar manufacturing facilities. Unknown metabolites can halt the time critical drug development process.
Liquid chromatography (LC) is a well-known, highly used and very valuable technique for the first line of mixture separation. A variety of methods and detectors can quantitatively and qualitatively analyze the contents eluted by the mobile phase, producing information on each component.
A single-wavelength UV–vis absorbance detector will provide a retention time for each band. Retention time relates to how long it takes for each component to travel the length of the column to the detector. This alone can provide a reasonable degree of diagnostic information because retention times are considered equivalent for compounds each time they are analyzed using the same stationary and mobile phase. More sophisticated analyses can be undertaken with detectors designed to provide more specific information. For example, photodiode-array (PDA) detectors monitor all wavelengths at once, which can be helpful in identifying components that are resolved incompletely by the chromatography process, provided that each component has a differentiating characteristic signal. Fluorescence, refractive index, radioisotope, electrochemical, and other detectors offer varying degrees of specificity and sensitivity when looking for particular compounds and compound classes. However, the ability of such detectors to assign an absolute structure to an unknown compound is limited to the extent that the combination of retention time and moiety detected (a chromophore, for example) can provide a sufficiently conclusive signature.
A significant step forward in the generation of more information-rich systems occurred in the 1970s, when the technical challenges of introducing a mobile phase, at atmospheric pressure, into the low pressure environment of a mass spectrometry (MS) system, were overcome. This capability proved immediately useful to many scientists (1) because the newly coupled technique of LC–MS could provide chromatographic and spectral information with more structural detail of a compound than most of the other available LC detectors. For example, the molecular weight of the compound as a whole and that of various substructure components are available. In addition, LC–MS provides visibility to a greater variety of compounds than just those with suitable chromophores, radiolabels, and so forth. Further, various MS techniques are available that institute disintegration of the component molecule into sequentially smaller pieces or fragments and track the various fragments, enabling a deeper understanding of each of the substructure components. One drawback of MS is that it is a destructive technique, and so it is typically used to sample small quantities of the mobile phase if it is desirable to retain the components of the mixture for further, separate analyses.
A roughly parallel development combined liquid chromatography with nuclear magnetic resonance (NMR) spectrometers to create an alternative information rich system (2,3). NMR is fundamentally the most structurally revealing detector available. It is capable of producing an atom-to-atom connectivity map of a molecule and creating a complete three-dimensional structure, thereby distinguishing between highly similar molecules, such as isomers. In addition, it is a nondestructive technique. Each component can be saved following the collection of NMR data. However, NMR is considerably less sensitive than MS, and only recently has the combination of advances in higher magnetic fields, probe designs, and instrument application techniques yielded sufficient sensitivity for LC–NMR to be truly viable for the groups that needed the structural detail afforded by this hyphenated system. Those groups predominantly study metabolites, natural products, impurities, degradants, and especially compounds known or suspected to have limited long-term stability, thereby requiring that separation be closely coupled to detection.
Many LC–NMR experiments can be run "on-flow." That is, the NMR instrument continuously records spectra as the mobile phase travels through the system. In addition, LC–NMR can use a UV–vis or PDA detector in line before the NMR (hence, the technique can be referred to as LC–UV–NMR to be aptly descriptive) to help make judicious choices of when the pump of the LC could be stopped, allowing a predetermined component to be held in stasis in the flow cell in the NMR system for long time-period data collection. This "stop-flow" technique allows the lack of sensitivity to be made up through extended acquisition, that is, signal averaging, and also allows more complex, more informative experiments to be run on components of interest.
The use of LC–NMR and, to a greater extent, LC–MS has become widespread within the pharmaceutical and chemical industries. Generally, an LC–MS system provides a fast, first screen that will filter out compounds that are more readily solved through library search techniques and fragment identification. The occasional and more intractable problems, resulting from complex, poorly understood molecular rearrangements or completely unknown impurities, will be run by LC–NMR. However, the time taken to solve the structure of an unknown is of critical importance, and so the use of an extended multihour NMR technique on the wrong component can be highly detrimental. This led to the development of a further "multiple hyphen" technique — LC–NMR-MS (4,5). This approach incorporates the advantages of both MS and NMR as information rich detectors for an LC system (see Figure 1). A split of the mobile phase, with 5% being directed to an MS instrument and 95% continuing to the NMR spectrometer, provides a means by which MS can be used to determine with greater specificity than UV–vis when the LC pump should be stopped for extended NMR interrogation of a particular peak or diversion of a particular peak into a loop collection or sample concentration system. In addition, triple-quadrupole or ion-trap MS devices can be used to generate fragment ion data on selected peaks. Now, long-term NMR experiments on samples of low concentration can be started automatically, with greater certainty that the correct compound is being studied.


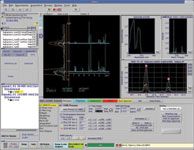
Figure 1: UV, MS, and NMR data, from two capsenoid compounds. 1H-NMR spectra of two LC peaks are stacked along with a vertical chromatogram trace. The two NMR data sets represented are capsaicin (9.46 min) and dihydrocapsaicin (10.98 min) confirmed both by NMR (absence of the methylene group at 5.2 ppm for the 10.98 min peak) and by MS, which shows a difference of two mass units between the two LC peaks.
In essence, an LC–NMR-MS system's general plumbing scheme is no more complex than that for a typical LC–MS or LC–NMR spectrometer. It is the performance and flexibility of the individual units and the degree of software integration that drives data acquisition, processing, viewing, and storage that is important. These factors enable the LC–NMR-MS system to become a practical working instrument.
Drug metabolites, impurities, and other compounds such as natural products (5,6) are generally available in very small quantities, so the performance of the NMR spectrometer, especially the sensitivity, is a key factor. Sensitivity, as well as being an intrinsic measure of the limit of detection, also determines instrument throughput because NMR is a signal averaging, Fourier-transform technique. Throughput measures the ability to generate NMR data on all components of interest within reasonable time frames, which is especially important because the UV and MS systems have two to three orders of magnitude better sensitivity than the NMR spectrometer and so will stand idle and are underutilized for the period that the NMR spectrometer is generating data during a stop-flow experiment. The use of cryogenically cooled flow probes (see Figure 2) generally has become accepted for these applications. Incorporating advanced cryogenic technologies to cool the electronics of the probe, and thereby reducing noise and providing substantial gains in sensitivity, these cold probes offer three to four times the sensitivity of conventional probes (a gain of 9–16-fold in throughput) (7).



Figure 2: Probes such as these can be used with both flow cells and conventional tubes. A standard NMR instrument can be converted to LCâNMR operation easily.
The solvents of choice for MS versus NMR can create practical problems that should be considered during method development for any sample. MS provides more instantly intuitive answers through the use of protonated solvents, while NMR prefers deuterated solvents. Deuterated solvents present the opportunity for proton–deuterium exchange, which generates problems when trying to interpret MS data. While the use of deuterated solvents can provide important information on the number of exchangeable protons in a sample, molecular ion information from that same sample is not immediately attributable to that for the all protonated species. By contrast, protonated water, methanol, and acetonitrile (commonly used solvents) produce intense signals in the 1H-NMR spectrum that can be reduced or eliminated by advanced solvent suppression techniques to yield the spectrum comprised mostly of the peaks of interest. A mobile phase containing deuterated solvents will generate little or no 1H signals and could be considered a superior solution because the suppression techniques also remove desirable signals that are masked by the protonated solvent signals. So, an NMR spectrometer that performs superior solvent suppression (that can precisely blank out the pinpoint ranges of the spectrum containing solvent peaks) is more greatly extendable to address a wider array of problems. A prime example is in rapid method development situations where working in protonated solvents typically provides faster, more cost-effective results — that is, more obvious data from the MS system and very suitable NMR spectra — without complex solvent exchange plumbing schemes.
For optimal productivity from a relatively complex system, software integration is perhaps the most obvious and necessary feature. Several important features determine how easily an LC–NMR-MS can be used for best advantage, including the ability for instruments to communicate with each other. Specifically, all four instruments, the LC, UV, MS, and NMR systems, should be addressable from one computer, and their methods all should be developed from one program that downloads all the separate methods to the respective devices for operation (see Figure 3). With one simple start command, the acquisition of data should be able to begin. To realize the complete benefit of stop-flow experiments, full flexibility is essential, and it is achievable with an instrument that has units that are able to communicate with each other during the course of the experiment. As described previously, the pump of the LC system is stopped and a peak of interest held in the NMR flow cell for extended periods. The criteria for stopping the flow can now be UV-, MS-, or MS-MS-based. UV triggers can typically be employed for compounds with specific retention times. However, MS or MS-MS can be used to trigger stop-flow NMR data acquisition on peaks that arise from components having certain molecular weights or molecular weight ranges, as well as from components that give rise to fragments of particular weights. The latter type of experiment is highly useful for metabolite or natural product analyses, where the molecular skeletons of a family of products can contain common fragments, making the search for the family members fairly straightforward. Once NMR data acquisition of a component of interest is complete, the NMR spectrometer signals the LC pump to restart and MS data acquisition resumes until another peak of interest dictates further stop–start cycles.


Figure 3: VnmrJ screen showing the Method Editor details for setting up an LCâNMR-MS experiment.
The bottom line is accessibility and usability of the data. To that end, a critical aspect of software integration is the ability to interactively view data from all three detectors — UV, MS, and NMR — and process data in a reasonably complete manner from one software package. This ensures that the full scale and richness of the information from all the detectors are completely accessible. Ideally, if a cursor is positioned at any given time point in a UV chromatogram, it should elicit the MS and NMR spectrum corresponding to the individual components. In addition, data resulting from more advanced MS-MS experiments, representing particular fragments, also should be easily viewable in context with both UV and NMR data. Often, NMR data are best viewed after postacquisition reprocessing techniques, such as phasing, baseline correction or other methods to improve data quality. To enable the full scope of structural elucidation capabilities of the system, these functions should be immediately available within the integrated software platform.
In conclusion, integrated, information rich detection systems, such as LC–NMR-MS, are providing multidimensional layers of knowledge critical to optimizing analyses and processes. They are opening avenues for researchers looking for creative ways to unravel challenging problems, particularly in the areas of metabolite and natural product identification. These tools and the data they generate are becoming increasingly accessible, giving researchers greater flexibility and creativity.
Iain Green is with Varian, Inc., Palo Alto, California.
References
(1) R. Willoughby, E. Sheehan, and S. Mitrovich, A Global View of LC/MS (Global View Publishing, Pittsburgh, Pennsylvania, 1998).
(2) N. Watanabe and E. Niki, Proc. Jpn. Acad., Ser. B, 54, 194–199 (1978).
(3) E. Bayer, K. Albert, M. Nieder, E. Grom, and T.J. Keller, J. Chromatogr. 186, 497–507 (1979).
(4) S.D. Taylor, B. Wright, E. Clayton, and I.D. Wilson, Rapid Commun. Mass Spectrom. 12, 1732–1736 (1998).
(5) J.P. Shockcor, S.E. Unger, I.D. Wilson, P.J.D. Foxall, J.K. Nicholson, and J.C. Lindon, Anal. Chem. 68, 4431–4435 (1996).
(6) K.I. Burton, J.R. Everett, M.J. Newman, F.S. Pullen, D.S. Richards, and A.G. Swanson, J. Pharm. Biomed. Anal. 15, 1903–1912 (1997).
(7) R.C. Crouch, W. Llanos, K. Mehr, C. Hadden, D.J. Russell, and G.E. Martin, Magn. Reson. Chem. 39, 555–558 (2001).












LIBS Illuminates the Hidden Health Risks of Indoor Welding and Soldering
April 23rd 2025A new dual-spectroscopy approach reveals real-time pollution threats in indoor workspaces. Chinese researchers have pioneered the use of laser-induced breakdown spectroscopy (LIBS) and aerosol mass spectrometry to uncover and monitor harmful heavy metal and dust emissions from soldering and welding in real-time. These complementary tools offer a fast, accurate means to evaluate air quality threats in industrial and indoor environments—where people spend most of their time.


Smarter Sensors, Cleaner Earth Using AI and IoT for Pollution Monitoring
April 22nd 2025A global research team has detailed how smart sensors, artificial intelligence (AI), machine learning, and Internet of Things (IoT) technologies are transforming the detection and management of environmental pollutants. Their comprehensive review highlights how spectroscopy and sensor networks are now key tools in real-time pollution tracking.

New AI Strategy for Mycotoxin Detection in Cereal Grains
April 21st 2025Researchers from Jiangsu University and Zhejiang University of Water Resources and Electric Power have developed a transfer learning approach that significantly enhances the accuracy and adaptability of NIR spectroscopy models for detecting mycotoxins in cereals.



